/home/virtual/kjpt/journal//../xmls/kjpt-45-3-254.xml
미토콘드리아 DNA 분석을 통한 구상나무와 분비나무의 계통지리학적 연구
적 요
분비나무와 구상나무의 계통지리적 유연관계 파악을 위하여 16개 지역의 구상나무와 분비나무 집단에 대하여 미토콘드리아 DNA(nad5 intron 4, nad5 intron 1 지역)를 이용한 유전적 분석을 수행하였다. 그 결과 총 7 지역의 유전자 변이가 확인되었으며, 4개의 반수체형이 확인되었다. 개체군 내 평균 유전다양성(HS)은 0.098, 전체 유전다양성(HT)은 0.620으로 관찰되었으며, 개체군 간 분화값은 GST = 0.841, NST = 0.849로 확인되었다. 조사 개체의 지리적 위치에 따라 일본지역을 제외하고 3개의 그룹(북부지역, 중부지역, 남부지역)으로 나누었다. 북부지역과 남부지역은 대부분 각각 M1, M2 단일의 반수체형을 가지며, 중부지역은 북부지역과 남부지역의 분포경계에 위치하면서 유전자 유입으로 인해 유전 다양성 (HT=0.654) 이 가장 높게 나타난 것으로 판단된다. 현재 남부지역의 단일의 반수체형(M2) 분포는 빙하기 때 북부지역에서 남하한 개체군들이 지리적 격리를 통해 분화하게 되고 빙하기 이후 다시 중부지역까지 분포 확장된 결과로 추측된다.
주요어: 분비나무, 구상나무, 계통지리, 미토콘드리아 DNA, 반수체형
Abstract
Genetic variations of Abies koreana and Abies nephrolepis were assessed using two mitochondrial DNA regions (nad5 intron 4 and nad5 intron 1) for 16 natural populations to understand their phylogeographical history. Seven polymorphic sites of the two combined regions resulted in the resolution of four haplotypes (M1−M4). The average gene diversity within the population (HS) was 0.098, the total gene diversity (HT) was 0.620, and the interpopulation differentiation was GST = 0.841, NST = 0.849. The populations were divided into three groups (northern area, central area, southern area) according to their geographic locations. The populations of the northern and southern areas were mostly fixed for M1 and M2, respectively. The populations of the central area showed the highest levels of gene diversity (HT = 0.654) due to introgression from the northern area and southern area. The presence of a single mtDNA haplotype in the southern area suggests that current widespread populations have expanded to the central area from a specific refugium population after the last glacial period.
Keywords: Abies koreana, Abies nephrolepis, phylogeography, mitochondrial DNA, haplotype
구상나무( Abies koreana Wilson, Pinaceae)는 한반도 특산 식물로서 제주도에서 덕유산까지 분포하는 것으로 알려져 왔다( Chung, 1958; Lee, 1996; Sun, 2007). 구상나무는 Wilson(1920)에 의해 근연분류군들과 비교하여 수피 및 수형, 수지구의 위치, 포의 형태 등의 특징으로 인해 새로운 종으로 발표되었다. 그러나 형질 변이의 연속성 및 다양성으로 인해 근연분류군인 분비나무[ Abies nephrolepis (Trautv. ex Maxim.) Maxim.]와 명확한 구분이 어려운 상황이다. 구상나무와 분비나무의 형질연구로는 Kim and Kim (1983)이 우리나라 전나무속( Abies)에 대한 전반적인 외부 및 해부형질에 대한 조사를 수행하였다. 그 후 Chang et al. (1997)은 수지구 위치의 비교 연구를 통해 지리산과 덕유산 지역에서 구상나무형과 분비나무형이 중복되어 나타나는 것으로 보고하였으며, 지리산 이남에 분포하는 개체들에서만 확인된 flavanone 성분이 중국의 A. faxoniana Rehder & E.H. Wilson [synonym of A. fargesii var. faxoniana (Rehder & E.H. Wilson) T.S. Liu.]과 A. georgei Orr [synonym of A. forrestii var. georgei (Orr) Farjon]에서 확인되어 이들 종이 구상나무의 원시종일 것으로 추정하였다. 또한 Han (2003)은 잎횡단면의 수지구 비교연구 결과 지역별로 중복 형질을 가지는 것으로 보고하였으며, Song et al. (2007)과 Song et. al. (2008)은 잎, 구과, 종자, 포침 등의 형질특성에 대한 연구에서 지역별 평균적 산출값의 비교를 통해 두 종이 구분되는 것으로 보고하였으나 개체군 내 중복형질은 명확히 구분되지는 않는 것으로 확인되었다. 소나무과의 식물은 피자식물과는 달리 엽록체 DNA가 부계유전하며, 미토콘드리아 DNA는 모계유전을 한다 ( Neale and Sederoff, 1989; Wagner et al., 1992). Abies속 식물들의 화분은 바람에 의해 산포되어 이동범위가 넓기 때문에 부계유전하는 엽록체 DNA 및 양성유전인 핵 DNA의 변이양상을 통해 지리적 유연관계를 파악하기에는 어려움이 있다( Burban and Petit, 2003; Du et al., 2009). 그렇기 때문에 최근에는 소나무과의 지리적 유연관계 및 계통학적 연구를 위하여 엽록체 DNA 및 핵 DNA와 함께 미토콘드리아 DNA가 많이 이용되고 있다( Wu et al., 1998; Liepelt et al., 2002; Ziegenhagen et al., 2005; Naydenov et al., 2007; Liepelt et al., 2010; Wang et al., 2011, Aizawa et al., 2012; Peng et al., 2012; Xiang et al., 2015). 따라서 본 연구에서는 미토콘드리아 DNA의 변이분석을 통해 구상나무와 분비나무의 지리학적 유연관계를 파악하고자 수행되었다.
재료 및 방법
DNA 추출, 증폭, 정제 및 염기서열결정
DNA 추출을 위해 확보한 재료의 잎을 Silica gel로 건조시켜 사용하였으며, 건조표본을 이용할 경우 가능한 최근에 수집된 표본의 어린잎을 사용하였다. 건조된 식물의 잎 40mg을 조직분쇄기 (Tissue Lyser II, QIAGEN, Germany)로 분쇄하여 1.5mL 튜브에 옮긴 후, DNeasy plant mini kit (QIAGEN, Germany)을 이용하여 공급자의 매뉴얼에 따라 Total genomic DNA를 추출하였다. 추출한 DNA는 0.9% agarose gel로 전기 영동하여 추출여부를 확인하였고, Nanodrop 2000 (Thermo scientific, USA)을 이용하여 DNA의 농도 및 순도를 측정하였다. 유전자 증폭을 위해 25−100 ng의 genomic DNA를 주형으로 Dr. MAX DNA polymerase (Doctor protein, Korea)를 이용하여 중합효소 연쇄반응을 진행하였다.
미토콘드리아 DNA의 nad5 intron 지역의 증폭에는 Wang et al. (2000)과 Liepelt et al. (2002)에 기존 연구에서 제시되었던 nad5 intron 1지역과 nad5 intron 4지역의 프라이머를 사용하였다( Table 2). 유전자 증폭을 위한 PCR과정은 95oC에서 5분 동안 pre-denaturation을 시킨 후, 95oC에서 30초의 denaturation, 58oC에서 30초의 annealing, 72oC에서 1분의 extension으로 이루어지는 thermal cycle 과정을 35회 반복하였으며, 마지막으로 72oC에서 7분간 final extension 과정을 진행하였다. PCR products는 multiscreen filter plate (Millipore Corp., USA)를 이용하여 공급자의 매뉴얼에 따라 정제하였으며, 정제된 PCR products는 BigDye® terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA)를 사용하여 cycle sequencing하였다. 염기서열의 분석은 ABI PRISM 3730XL Analyzer (Applied Biosystems, USA)를 이용하였다. 이를 통해 nad5 intron 1 유전자의 경우 총 266개, nad5 intron 4 유전자의 경우 총 304개의 염기서열을 확인하였다. 각 지역의 염기서열들은 모두 NCBI의 GenBank에 등록되었으며 accession number를 부여받았다(KT448846−KT449415).
데이터 분석
본 연구 결과의 반수체형 네트워크(haplotype network) 분석을 위하여 TCS ver. 1.21 ( Clement et al., 2000)을 사용하였으며, 개체군 내 유전다양성( H; Nei, 1987)은 ARLEQUIN ver. 3.5.2.1 ( Excoffier et al., 2005)을 사용하여 분석하였다. 개체군 내 유전다양성 평균지수( HS)와 총 유전다양성지수( HT) 그리고 개체군 간 분화지수( GST와 NST)는 PERMUT ver. 1.2.1을 사용하여 분석하였다(used 1,000 permutations). GST 값은 대립형질(allele)의 빈도를 기반으로 하며, NST 값은 haplotype의 빈도와 haplotype들 사이의 유사성에 의해 계산된다. 만약 NST값이 GST값 보다 높으면 유연 관계가 높은 haplotype들이 특정 개체군 내에서 함께 발생되었을 가능성이 높은 것을 의미하며, 분석된 개체군들은 유의한 계통지리학적 구조(phylogeographical structure)를 가지는 것으로 판단할 수 있다( Pons and Petit, 1996). 계층적으로 개체군의 유전적 변이를 확인하기 위한 AMOVA (Analysis of molecular variance) 분석은 ARLEQUIN에 의해 수행되었다. 개체군들의 지리적 비교 분석을 위하여 분비나무 집단으로 생각되는 경기·강원도 이북 (위도 37o이상)을 북부, 구상나무 집단으로 생각되는 전라도 이남 (위도 36o이하)을 남부, 그리고 두 분류군의 중복 분포가 나타나는 충청도 (위도 36o−37o)지역을 중부지역으로 3개의 그룹으로 나누어 구분하였다(일본지역은 개체 수가 적어 개체군 분석에서 제외하였음).
결 과
mtDNA의 변이와 염기서열 분포
구상나무와 분비나무의 유전적 차이를 확인하기 위하여 미토콘드리아 DNA 2지역( nad5 intron 4, nad5 intron 1)에 대해 집단별 염기서열 분석을 수행하였다. 그 결과 nad5 intron 4지역의 염기서열은 298−302 bp, nad5 intron 1지역은 1,259 bp가 확인되었으며, 총 7개의 변이 지역이 확인되었다( Table 3).
nad5 intron 4 에서는 191−194 bp에서 1개의 indel(insertion/deletion)이 확인되었으며, 그 외 5개의 단일 염기서열(38, 187, 220, 222, 238 bp)에서 치환현상이 관찰되어 총 6개의 변이 지역이 확인되었다. nad5 intron 1 에서는 단일염기서열(543 bp)의 치환현상으로 인해 1개의 변이 지역이 확인되었다. 확인된 변이 지역에 의해 4개의 mtDNA haplotype (mitotypes)이 선정되었다( Table 3). 북부지역에서는 Mitotype M1과 M2가 확인되었으며, 중부지역에서는 M1, M2, M3, 남부지역에서는 M2, M3가 확인되었고, 일본의 Abies veitchii에서 M4가 확인되었다. Mitotype M1은 백두산지역에서 부터 강원도 함백산지역까지 관찰되었으며, 전형적인 분비나무형으로 판단된다. 가리왕산지역에서는 M2형을 갖는 개체가 일부 확인되었다. M2는 M1과 비교하여 1개의 indel과 2개의 염기서열 치환에 의한 차이가 있다. 소백산지역에서 부터 한라산지역까지 확인되었으며, 전형적인 구상나무형으로 판단된다. M3는 중부지역의 월악산, 속리산과 남부지역의 영축산 지역에서 관찰되었다( Fig. 1A).
Mitotype network 분석에서는 M1으로 부터 1개의 indel이 발생한 후 M2 그룹과 M3와 M4 그룹 두 개로 크게 나뉘어졌다. M2와 비교하여 M3는 6개의 유전자 변이 지역이 확인되었으며, M4는 5개의 변이 지역이 확인되었다( Fig. 1B).
mtDNA에 의한 유전다양성
mtDNA를 통한 구상나무와 분비나무의 유전다양성 분석 결과 개체군별로는 가리왕산( H = 0.466), 소백산( H = 0.372), 속리산( H = 0.333), 월악산( H = 0.303) 순으로 나타났다 ( Table 1). 개체군 내 평균 유전다양성( HS)은 0.098, 전체 유전다양성( HT)은 0.620로 관찰되었다. 개체군 간 분화값은 GST = 0.841, NST = 0.849로 확인되었으며, NST 값이 GST값에 비해 높게 관측되어 유의적 계통지리학적 구조를 가지는 것으로 나타났다. 지역별로는 북부지역이 GST = NST = 0.222로 다른 지역에 비해 다소 낮게 확인되었고, 중부지역에서는 GST = 0.486, NST = 0.503으로 관찰되어 북부지역에 비해 개체군 간 분화값이 높은 것으로 확인되었다. 남부지역에서는 개체군 내 유전다양성( HS= 0)이 관측되지 않았으며, 그 결과 개체군 간 분화값은 GST = NST = 1.000 으로 관찰되었고, 전체 유전다양성( HT)은 0.286으로 북부지역보다는 높고 중부지역 보다는 낮은 것으로 측정되었다( Table 4). AMOVA (Analysis of molecular variance) 분석에서는 그룹 간 변이가 71.88%로 가장 크게 관찰되었으며, 그룹 내 개체군 간 변이는 14.74%가 확인되었고, 개체군 내 변이는 13.38%로 가장 낮게 나타났다 ( Table 5).
고 찰
유전적 다양성과 분화
지역별로는 북부지역에서 남부지역으로 개체 간 분화가 더 높게 일어났다(GST 0.222<0.486<1.000). 남부지역의 높은 유전분화값은 남서쪽의 M2와 남동쪽의 M3 개체군들 사이의 뚜렷한 분화에 의해 나타난 현상으로 판단된다. 중부지역은 개체군 내 유전다양성(HS = 0.336)과 전체 유전다양성(HT = 0.654)이 가장 높게 관찰되었다. 중부지역의 경우 유일하게 3개의 haplotype (M1, M2, M3)이 확인되었다. 이는 중부지역이 북부지역과 남부지역의 경계지역으로서 두 지역에서 나타나는 개체군 유형이 중복되어 분포하기 때문인 것으로 판단된다. 북부지역의 경우 가리왕산(H = 0.466)을 제외한 다른 집단의 유전다양성(H)은 0으로 확인되었으며, 이는 중부지역으로부터 지리적으로 가까운 가리왕산 집단에 종자산포로 인한 유전자의 역유입이 일어난 것으로 판단된다.
Mitotype network 분석 결과에서 M1으로 부터 1개의 indel이 발생한 후 M2 그룹과 M4 그룹 두 개로 크게 나뉘어졌으며, M4로 부터 다시 M3가 분화되는 것으로 확인되었다. 이는 일본지역까지 남하한 분비나무 혹은 조상종이 유전분화 이후 분포확장을 통해 한반도로 재유입되었을 가능성을 나타내준다.
계통지리학적 분석
Abies속 식물은 북반구 지역을 중심으로 하여 전 세계 약 40여종이 분포한다( Farjon, 1990). 최초의 Abies속 식물들 화석은 Eocene(56−33.9 백만년 전) 시기에 북아메리카와 동아시아지역에서 확인되었다. 특히 아시아지역에 현존하는 대부분의 Abies속 식물들은 서로간의 유전적 분화가 낮아 비교적 최근에 (Pliocene−Pleistocene, 5.3−0.01백만년) 분화된 것으로 알려져 있다( Semerikova and Semerikov, 2014). 한반도 Abies속의 분포는 화석자료를 바탕으로 하여 주로 신생대 3기 Miocene (23 백만년 전) 부터 한반도 전역에 걸쳐 확인되고 있다( Kong, 2004),
현존하는 한반도 Abies속은 모두 3종( A. koreana, A. nephrolepis, A. holophylla)으로 주로 해발 1,000 m 이상의 고산지대에 주로 분포하고 있다. 한랭성의 Abies속 식물들은 여러 번의 빙하기를 거치면서 남부지역으로 분포의 확장과 축소를 반복하였으며, 그 결과 빙하기 때 내려온 개체들이 빙하기 이후 온도가 상승함에 따라 고산지역에 남아있게 된 것으로 알려지고 있다( Hong et al., 2011). 형태적으로 유사한 분비나무와 구상나무는 한반도 내 분포가 서로 접해있으며, 그 경계에 있는 개체들은 형태적으로 서로 중복형질을 갖게 되어 명확한 구분이 어려운 상황이다.
본 연구에서 구상나무와 분비나무 개체들의 mtDNA 분석결과 북부지역 개체군들과 남부지역의 개체군 들은 서로 다른 단일의 haplotype (M1, M2)을 가지면서 지리적 구분이 이루어졌다. 일반적으로 종의 유전적 분화는 지리적 격리에 의해 이루어지나( Meng et al., 2007), 한반도 중부지역에서 특별한 지리적 격리 기작은 확인되지 않는다. 다른 한편으로 넓은 분포에 걸쳐 단일의 유전자형을 갖는다는 것은 어느 특정지역(refugium 등)으로 부터의 분포확장에 의한 것일 가능성이 높다( Avise, 2004; Du et al., 2009). Aizawa et al. (2012)은 중국, 한국, 일본에 분포하는 잣나무( Pinus koreaiensis)들의 mtDNA 분석결과 대륙의 잣나무들은 모두 단일의 유전자형을 갖는 반면 일본의 잣나무들은 보다 다양한 유전자형을 갖는 것으로 확인되어 대륙의 잣나무들이 빙하기 때 분포가 축소되었다가 빙하기 이후 남부지역으로 부터의 분포확장에 의해 구성된 것으로 판단하고 있다. 이를 근거로 중부지역 단일 유전자형(M2)은 빙하기에 북부지역에서 남하한 분비나무 혹은 분비나무의 조상종이 한라산지역까지 분포지가 확장되고, 이후 한라산지역에서 유전적 고립에 의해 M2의 유전형으로 분화되었으며, 빙하기 이후 온도상승에 따라 한라산지역의 M2 개체들이 중부지역까지 분포확장된 결과로 생각된다. 구상나무의 cpDNA 분석 연구( Kormutak et al., 2007)에서는 덕유산( H = 0.205)과 지리산 개체군( H = 0.167)과 비교하여 한라산 개체군의 유전다양성이 가장 높은 것( H = 0.21)으로 확인되었으며, 한라산지역이 오랜기간에 걸쳐 다른 지역 보다 유전분화가 더욱 심화된 것으로 판단된다. 또한 북부지역에 M1과 남부지역의 M2의 분포경계가 중부지역의 형성되면서 중부지역 유전다양성( HT = 0.654)이 높아진 것으로 판단된다. 최대 빙하기 때(Last glacial maximum, LGM, 약 2만년 전) 한반도의 동, 서지역은 기후적인 차이가 있었다( Yoon and Hwang, 2009). 서부지역은 해수면이 낮아지면서 서해지역이 육지화되고 매우 건조하였으며, 동부지역에 비해 다소 온난한 기후를 가졌을 것으로 알려졌다. 또한 목본식물의 분포에 있어 동해안지역에는 전나무속, 잣나무속, 가문비나무와 자작나무가 우점하였으며, 서해안 지역에는 소나무속, 잣나무속, 참나무속이 우점하였을 것으로 예상되어지고 있다. 본 연구에서 확인된 남동지역의 haplotype M3 개체들은 최대빙하기 때 동, 서부지역의 기후적 차이로 인하여 M2 개체들과는 별개의 유전분화 및 분포확장이 이루어진 것으로 생각되어진다. 신생대 4기 초와 중기(30−120만 년 전)에 형성된 육로( Dobson and Kawamura, 1998)를 통해 일본지역으로 유입된 분비나무 혹은 분비나무 조상종은 지리적 격리로 인해 유전적 분화(M4)가 일어나고, 최대 빙하기 때 해수면이 낮아지면서 한반도와 일본 사이 대한해협(Korea Strait)이 다시 육지로 연결되면서( Kimura, 2000; Yoon and Hwang, 2009; Kim et al., 2010) 한반도로 재유입되었을 것으로 생각된다. 한반도로 유입된 M4 개체들은 한반도 남동지역에서 M3로 유전분화가 일어나고 중부지역까지 분포가 확장되었을 것이다. 본 연구는 미토콘드리아 DNA 일부 구간만을 대상으로 수행되었기에 분비나무와 구상나무의 명확한 지리학적 유연관계를 판단하기에는 부족함이 있다. 그러나 한반도에 분포하는 Abies속 개체들에 대하여 우리나라 최초로 모계유전하는 미토콘드리아 DNA 정보를 확인하고, 이와 함께 전반적인 지리적 유연관계 파악이 이루어졌다는 점에서 의미가 있는 것으로 판단된다. 분비나무와 구상나무의 보다 심도있는 계통지리학적 분석을 위해서는 핵 DNA, 엽록체 DNA 등 더욱 다양한 유전구간과의 비교 검토와 함께 세부적인 형태특성 연구가 필요할 것으로 생각된다.
Fig. 1.
Geographical distributions (A) and network (B) of mtDNA haplotypes (mitotypes) of Abies koreana and Abies nephrolepis (A. veitchii in japan). In the network, solid bar (single nucleotide substitution) and open bar (indel) on the branches represent mutational steps. 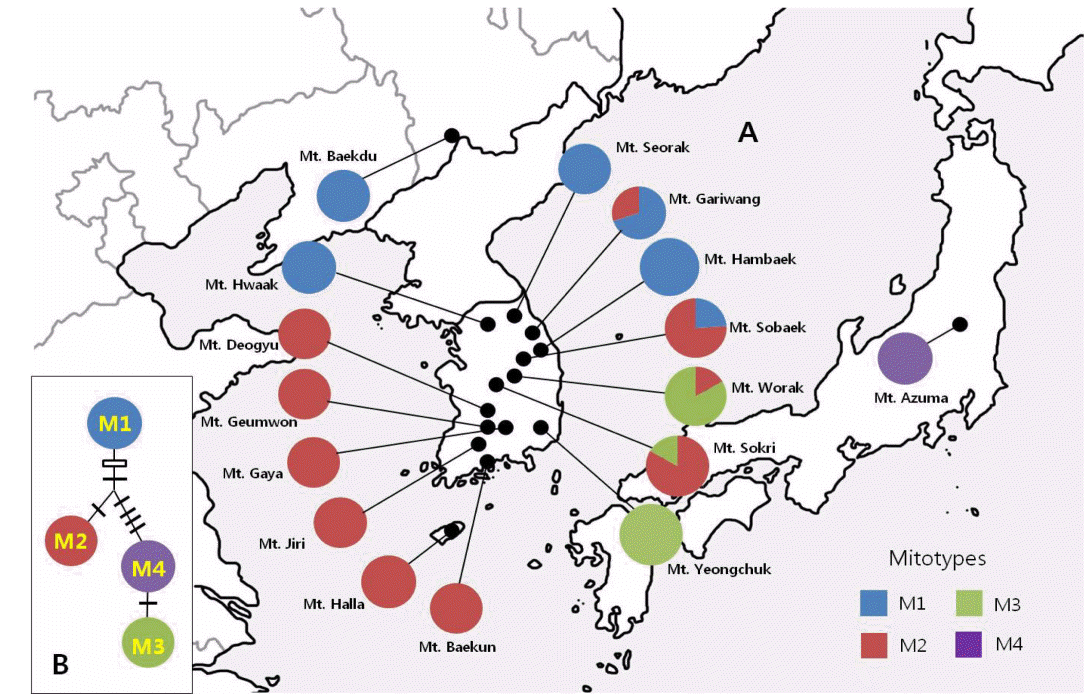
Table 1.
Locations of Abies koreana and A. nephrolepis, proportion of individuals exhibiting each mitotype per population, and gene diversity (H) values for mitotypes.
|
Taxon |
Population |
Country |
Lat. |
Long. |
N
|
Mitotype
|
H
|
|
M1 |
M2 |
M3 |
M4 |
|
Abeis nephlorephis and A. koreana complex |
Northern area |
|
|
|
|
|
|
|
|
|
|
Mt. Baekdu |
China |
42° 04' |
128° 03' |
31 |
31 |
0 |
0 |
0 |
0 |
|
Mt. Seorak |
Korea |
38° 07' |
128° 27' |
10 |
10 |
0 |
0 |
0 |
0 |
|
Mt. Hwaak |
Korea |
37° 59' |
127° 30' |
8 |
8 |
0 |
0 |
0 |
0 |
|
Mt. Gariwang |
Korea |
37° 28' |
128° 34' |
10 |
7 |
3 |
0 |
0 |
0.466 |
|
Mt. Hambaek |
Korea |
37° 09' |
128° 55' |
25 |
25 |
0 |
0 |
0 |
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Central area |
|
|
|
|
|
|
|
|
|
|
Mt. Sobaek |
Korea |
36° 57' |
128° 28' |
50 |
12 |
38 |
0 |
0 |
0.372 |
|
Mt. Worak |
Korea |
36° 53' |
128° 06' |
12 |
0 |
2 |
10 |
0 |
0.303 |
|
Mt. Sokri |
Korea |
36° 33' |
127° 52' |
6 |
0 |
5 |
1 |
0 |
0.333 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Southern area |
|
|
|
|
|
|
|
|
|
|
Mt. Deogyu |
Korea |
35° 51' |
127° 44' |
19 |
0 |
19 |
0 |
0 |
0 |
|
Mt. Gaya |
Korea |
35° 49' |
128° 07' |
11 |
0 |
11 |
0 |
0 |
0 |
|
Mt. Geumwon |
Korea |
35° 43' |
127° 45' |
8 |
0 |
8 |
0 |
0 |
0 |
|
Mt. Jiri |
Korea |
35° 19' |
127° 41' |
20 |
0 |
20 |
0 |
0 |
0 |
|
Mt. Baekun |
Korea |
35° 06' |
127° 37' |
13 |
0 |
13 |
0 |
0 |
0 |
|
Mt. Halla |
Korea |
33° 22' |
126° 33' |
35 |
0 |
35 |
0 |
0 |
0 |
|
Mt. Yeongchuk |
Korea |
35° 31' |
129° 03' |
5 |
0 |
0 |
5 |
0 |
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Abies veitchii
|
Mt. Azuma |
Japan |
37° 41' |
140° 54' |
3 |
0 |
0 |
0 |
3 |
0 |
Table 2.
Primer sequences for analyzing the mtDNA of Abies.
|
Gene |
Label |
Sequence |
Reference |
|
1 |
nad5 intron 1 |
F |
nad5-aF |
GGAAATGTTTGATGCTTCTTGGG |
Wang et al. (2000) |
|
R |
nad5-bR |
CTGATCCAAAATCACCTACTCG |
|
2 |
nad5 intron 4 |
F |
nad5-4F |
CATCCCTCCCATTGCATTAT |
Liepelt et al. (2002) |
|
R |
nad5-4R |
GGACAATGACGATCCGAGATA |
Table 3.
Variable sites of the aligned sequences of two mtDNA fragment in four haplotypes.
|
Mitotype |
nad5 intron 4
|
nad5 intron 1
|
|
38 |
187 |
191-194 |
220 |
222 |
238 |
543 |
|
M1 |
G |
C |
A |
T |
A |
T |
G |
T |
T |
C |
|
M2 |
G |
A |
- |
- |
- |
- |
G |
T |
T |
A |
|
M3 |
T |
C |
- |
- |
- |
- |
T |
G |
G |
C |
|
M4 |
T |
C |
- |
- |
- |
- |
T |
G |
G |
A |
Table 4.
Estimates of average gene diversity within population (HS), total gene diversity (HT), and two measures of interpopulation differentiation (GST and NST) (mean ± SE in parentheses) for mitotypes.
|
Areas |
HS
|
HT
|
GST
|
NST
|
|
Northern area |
0.093 (0.093) |
0.120 (0.105) |
0.222 (NC) |
0.222 (NC) |
|
Central area |
0.336 (0.020) |
0.654 (0.097) |
0.486 (0.126) |
0.506 (0.121) |
|
Southern area |
0.000 (0.000) |
0.286 (0.204) |
1.000 (NC) |
1.000 (NC) |
|
Whole area |
0.098(0.444) |
0.620(0.069) |
0.841(0.073) |
0.849(0.070) |
Table 5.
Analysis of molecular variance (AMOVA) data for Abies koreana and A. nephrolepis populations from three geographical regions in Korea.
|
Source of variation |
d.f.
|
SS |
VC |
Variation (%) |
Fixation index |
P value |
|
Among areas |
2 |
54.19 |
0.263 |
71.88 |
FCT=0.524 |
<0.001 |
|
Among populations within areas |
12 |
11.15 |
0.054 |
14.74 |
FSC=0.866 |
<0.001 |
|
Within populations |
280 |
13.72 |
0.049 |
13.38 |
FST=0.718 |
<0.001 |
|
Total |
294 |
79.06 |
0.366 |
100 |
|
|
Literature Cited
Aizawa, M. Kim, S and Yoshimaru, H. 2012. Phylogeography of the Korean pine ( Pinus koraiensis) in northeast Asia: inferences from organelle gene sequences. Journal of Plant Research 125(6): 713-723.   Avise, JC. 2004. Molecular markers, natural history, and evolution (Second edition). Sunderland, MA, Sinauer.
Burban, C and Petit, J. 2003. Phylogeography of maritime pine inferred with organelle markers having contrasted inheritance. Molecular Ecology 12(6): 1487-1495. Chang, CS. Jeon, I and Hyun, O. 1997. An analyasis of morphological variation in Abies koreana Wilson and A. nephrolepis (Traut.) Maxim. of Korea (Pinaceae) and their phylogenetic problems. Journal of Korean Forestry Society 86: 378-390 (in Korean).
Chung, T.H. 1958. Korean Flora. Vol. 1. Woody plants. Shinjisa, Seoul. (in Korean).
Clement, M. Posada, D and Crandall, K. 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9(10): 1657-1660. Dobson, M and Kawamura, Y. 1998. Origin of the japanese land mammal fauna: allocation of extant species to historically-based categories. The Quaternary Research 37(5): 385-395. Du, F.K. Petit, J and Liu, Q. 2009. More introgression with less gene flow: chloroplast vs. mitochondrial DNA in the Picea asperata complex in China, and comparison with other conifers. Molecular Ecology 18(7): 1396-1407. Excoffier, L. Laval, G and Schneider, S. 2005. Arlequin var. 3.0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online 1: 47-50.  Farjon, A. 1990. Pinaceae: drawings and descriptions of the genera Abies, Cedrus, pseudolarix, Keteleria, Nothotsuga, tsuga, Cathaya, Pseudotsuga, Larix and Picea. Konigstein. Koeltz Scientific Books,
Han, TI. 2003. An anatomical tudy of cross section needles of Abies koreana and A. nephrolepis. MS thesis. Kong Ju National University. (in Korean).
Hong, Y.P. Ahn, Y. Kim, M. Yang, H and Son, H. 2011. Genetic variation of nSSR markers in natural populations of Abies koreana and Abies nephrolepis in South Korea. Journal of Korean Forestry Society 100(4): 577-584 (in Korean).
Jaramillo-Correa, JP. Aquirre-Planter, E. Khasa, P. Equiarte, E. Piñero, D. Furnier, R and Bousquet, J. 2008. Ancestry and divergence of subtropical montane forest isolates: molecularmolecular biogeography of the genus Abies (Pinaceae) in southern México and Guatemala. Molecular Ecology 17(10): 2476-2490. Jung, YH. Han, H. Oh, S. Ko, H. Koh, C. Kim, H and Oh, Y. 2000. Genetic variation and phylogenetic analysis of genus Abies Distributed in Korea based on RAPD and nuclear rDNA ITS2 sequences. Korean Journal of Genetics 22(4): 341-354 (in Korean).
Kim, IS and Hyun, O. 2000. Genetic diversity of Abies koreana Wilson based on RAPD analysis. Korean Journal of Breeding Science 32(1): 12-18 (in Korean).
Kim, JM. Kennett, P. Park, K. Kim, C. Kim, Y and Roark, B. 2010. Paleoceanographic change during the last deglaciation, East sea of Korea. Paleoceanography 15(2): 254-266. Kim, YD and Kim, S. 1983. Studies on the Morphological and anatomical characteristics of genus Abies in Korea. Journal of Korean Forestry Society 62: 68-75 (in Korean).
Kimura, M. 2000. Paleogeography of the Ryukyu islands. Tropics 10(1): 5-24. Kong, WS. 2004. Species composition and distribution of native korean conifers. Journal of the Korean Geographical Society 39(4): 528-543.
Kormutak, A. Hong, P. Kwon, Y and Kim, S. 2007. Variation in trn-L/trn-V and trn-F/trn-T spacer regions of cpDNA in Abies koreana Wilson and A. nephropepis Traut./Maxim. Journal of Korean forest Society 96(2): 131-137.
Lee, SW. Yang, H. Han, D. Song, H and Lee, J. 2008. Genetic variation in natural populations of Abies nephrolepis Max. in South Korea. Annals of forest Science 65: 302. Lee, W.T. 1996. Coloured standard illustrations of korean plant. Academy Publishing Co, Seoul. (in Korean).
Liepelt, S. Mayl, E. -Quellhorst, . Lahme, M and Ziegenhagen, B. 2010. Contrasting geographical patterns of ancient and modern genetic lineages in mediterranean Abies species. Plant Systematics and Evolution 284: 141-151. Liepelt, S. Bialozyt, R and Ziegenhagen, B. 2002. Wind-dispersed pollen mediates postglacial gene flow among refugia. Proceedings of the National Academy of Sciences 99(22): 14590-14594. Meng, L. Yang, R. Abbott, J. Miehe, G. Hu, T and Liu, J. 2007. Mitochondrial and chloroplast phylogeography of Picea crassifolia Kom. (Pinaceae) in the Qinghai-Tibetan plateau and adjacent highlands. Molecular Ecology 16: 4128-4137. Neale, DB and Sederoff, RR. 1989. Paternal inheritance of chloroplast DNA and maternal inheritance of mitochondrial DNA in loblolly pine. Theoretical and Applied Genetics 77: 212-216. Nei, M. 1987. Molecular evolutionary genetics. Columbia University Press, New York.
Peng, Y. Yin, S. Wang, J. Tian, B. Ren, G. Guo, Q and Liu, J. 2012. Phylogeographic analysis of the fir species in southern China suggests complex origin and genetic admixture. Annals of Forest Science 69(3): 409-416. Pons, O and Petit, RJ. 1996. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics Society of America 144: 1237-1245.
Semerikova, SA and Semerikov, VL. 2014. Molecular phylogenetic analysis of the genus Abies (Pinaceae) based on the nucleotide sequence of chloroplast DNA. Russian Journal of Genetics 50(1): 7-19. Song, JH. Lee, JJ and Kang, KS. 2008. Variation in cone, seed, and bract morphology of Abies nephrolepis (Trautv.) Maxim. and A. koreana Wilson in native forests. Journal of Korean Forestry Society 97: 565-569 (in Korean).
Song, JH. Lee, J. Lee, Y. Lee, C and Kim, Y. 2007. Variation in needle morphology of natural populations of Abies nephrolepis Maxim. and A. Koreana Wilson in Korea. Journal of Korean Forestry Society 96: 387-392 (in Korean).
Sun, YB. 2007. Pinaceae in the genera of vascular plants of Korea. flora of Korean Editorial committee (eds.). Academy Puyblishing Co., Seoul. 119 pp.
Wagner, DB. 1992. Nuclear, chloroplast and mitochondrial DNA polymorphisms as biochemical markers in population genetic analysis of forest trees. New Forests 6: 373-390. Wang, XQ. Tank, C and Sang, T. 2000. Phylogeny and divergence times in Pinaceae: evidence from three genomes. Molecular Biology and Evolution 17(5): 773-781. Wilson, EH. 1920. Four new conifers from Korea. Journal of the Arnold Arboretum 1: 186-190.
Wu, J. Krutovskii, KV and Strauss, SH. 1998. Abundant mitochondiral genome diversity, population differentiation and convergent evolution in Pines. Genetics Society of America 150(4): 1605-1614.
Xiang, QP. Wei, R. Shao, Z. Yang, Y. Wang, Q and Zhang, C. 2015. Phylogenetic relationships, possible ancient hybridization, and biogeographic history of Abies (Pinaceae) based on data from nuclear, plastid, and mitochondrial genomes. Molecular Phylogenetics and Evolution 82: 1-14. Yoon, SO and Hwang, I. 2009. The Resconstruction of natural environment for the last glacial maximum around Korea and adjacent area. Jurnal of the korean geomorphological association 16(3): 101-112.
Ziegenhagen, B. Fady, B. Kuhlenkamp, V and Liepelt, S. 2005. Differentiating groups of Abies species with a simple molecular marker. Silvae Genetica 54(3): 123-126.
|
|











