Phylogenetic analysis of Neottia japonica (Orchidaceae) based on ITS and matK regions
Article information

Abstract
To elucidate the molecular phylogeny of Neottia japonica, which is a terrestrial orchid distributed in East Asia, the internal transcribed spacer (ITS) of nuclear DNA and the matK of chloroplast DNA were used. A total 22 species of 69 accessions for ITS and 21 species of 114 accessions for matK phylogeny were analyzed with the maximum parsimony and Bayesian methods. In addition, we sought to establish a correlation between the distribution, morphology of the auricles and genetic association of N. japonica with phylogenetic data. The phylogenetic results suggest that N. japonica is monophyletic and a sister to N. suzukii in terms of the ITS phylogeny, while it is paraphyletic with N. suzukii in terms of the matK phylogeny. N. japonica and N. suzukii show similar morphologies of the lip and column, they both flower in April, and they are both distributed sympatrically in Taiwan. Therefore, it appears to be clear that N. japonica and N. suzukii are close taxa within Neottia, although there is incongruence between the nrDNA and cpDNA phylogenies of N. japonica. The incongruence between the two datasets may have various causes, meaning that further studies are needed to confirm the evolutionary process of N. japonica. The phylogenetic status of N. kiusiana, which was not included in previous studies, was as a sister to N. nidus-avis. Meanwhile, the ITS and matK phylogenies are unsuitable for identifying genetic associations with the characteristic of auricles. The phylogenetic topologies of Korean, Taiwanese and mainland Chinese individuals suggest that the populations of N. japonica in Korea originated from China’s mainland and island areas. The characterization of regional gene differences could provide useful preliminary data for future studies.
The genus Neottia Guett. belongs to the Neottieae tribe of the Orchidaceae (Dressler, 1993; Cameron et al., 1999; Chase et al., 2003) and is comprised by approximately 60–70 taxa distributed throughout eastern and northern Asia, Europe, and North America, with a few species extending into tropical Asia (Pridgeon et al., 2005; Chen et al., 2009). More than 70% of Neottia species are found in East Asia, making this region a diversity hotspot for this genus. Nevertheless, few phylogenetic studies have focused on this genus and relatively few species have been characterized (Xiang et al., 2012; Feng et al., 2016; Zhou and Jin, 2018).
The genus Neottia includes both autotrophic and mycoheterotrophic plants. Most autotrophic plants possess two opposite leaves (sometimes three or more) in the middle of the stem. On the other hand, mycoheterotrophic plants are characteristically achlorophyllous and possess densely fleshy bird-nest-like roots. These distinct morphological differences and trophic types formerly divided the genus into Listera and Neottia (Bentham, 1881; Pfitzer, 1887; Schlechter, 1926; Brieger et al., 1974; Dressler, 1981; Rasmussen, 1982). However, more recent DNA analyses identified Listera as a photosynthetic group of Neottia, and therefore suggested merging the two genera (Chase et al., 2003; Bateman et al., 2005; Chen et al., 2009).
Another systematic molecular study of the Neottieae tribe by Zhou and Jin (2018) elucidated a new phylogenetic relationship within the genus Neottia, as well as the evolution of mycoheterotrophic orchids. This study demonstrated that the molecular features of Neottia considerably correlated with its morphological characteristics and distribution. Moreover, mycoheterotrophic orchids appear to have evolved independently from their autotrophic counterparts within the genus Neottia. The phylogeny of N. japonica was recently elucidated for the first time (Zhu et al., 2019). However, only eight Neottia species were included in this study, and although these studies identified new phylogenetic relationships among Neottia and N. japonica, additional studies encompassing a wider range of taxa are required.
Neottia japonica (Blume) Szlach. is an autotrophic terrestrial orchid found in warm-temperate regions of Japan, Taiwan, China, and Korea (Su, 2000; Yu and Xiang, 2009; So et al., 2013; Liu et al., 2014; Ohashi, 2015; Zou et al., 2018). It is distributed in Honshu, Shikoku, Kyushu, and the Ryukyu archipelagos of Japan (Ohashi, 2015), as well as in the northern Taiwan forests at altitudes of 1,400–3,000 m (Su, 2000). In Korea, N. japonica is restricted to the southern part of Jejudo Island. In China, N. japonica has been identified in Hunan (Yu and Xiang, 2009); however, according to the authors, its current distribution in this region remains unclear. More recently, the species was reported in Zhejiang (Liu et al., 2014) and Guangxi (Zou et al., 2018). As such, the distribution of this species has been characterized relatively recently.
The morphological characters that distinguish N. japonica from related taxa are its T-shaped lamella and elongated auricles embracing the column of the lip. Moreover, Neottia species native to Korea bloom from May to August, whereas N. japonica blooms in April (Lee, 2011; So et al., 2013). The shape of the auricles at the floral portions varied in the populations from Korea, Taiwan, and Japan. The Japanese plants possess auricles that are long enough to overlap one another, whereas the auricles of Korean and Taiwanese plants are too short to overlap. Auricle images are presented in Fig. 1.

Morphological distinguish point at the floral part (A–E). A. Korean type-short type. B. Japanese type-long type. C. Taiwanese typeshort type. D. Illustration of lip part (So et al., 2013). The arrows indicate tip of auricles. E. Habit (pictured from Jejudo Island of Korea).
Therefore, the aims of this study were (1) to elucidate the molecular phylogeny of Neottia japonica and its alliance species, and (2) to infer the phylogenetic status of N. kiusiana, (3) to establish a correlation between the distribution, morphological characteristics and genetic association of N. japonica collected from East Asia.
Materials and Methods
Plant sampling
Leaf samples were collected from Korea, Japan and Taiwan. The localities are pointed on the map (Fig. 2). Leaves used as DNA sources were collected from the natural populations. Fresh or dried leaves were used to extracted total DNA. Plant samples were collected apart from each individual for avoiding colony with same rhizome. Voucher information of plant materials, GenBank accession numbers and previous sequence data by Zhou and Jin (2018) are listed in Table 1. For internal transcribed spacer (ITS) of nuclear DNA (nrDNA below) phylogenetic analysis, 69 accessions from 22 taxa of Neottia. For a chloroplast DNA (cpDNA below) phylogeny, 114 accessions including 21 taxa of Neottia. All voucher specimens were deposited at Ewha Womans University Herbarium (EWH).
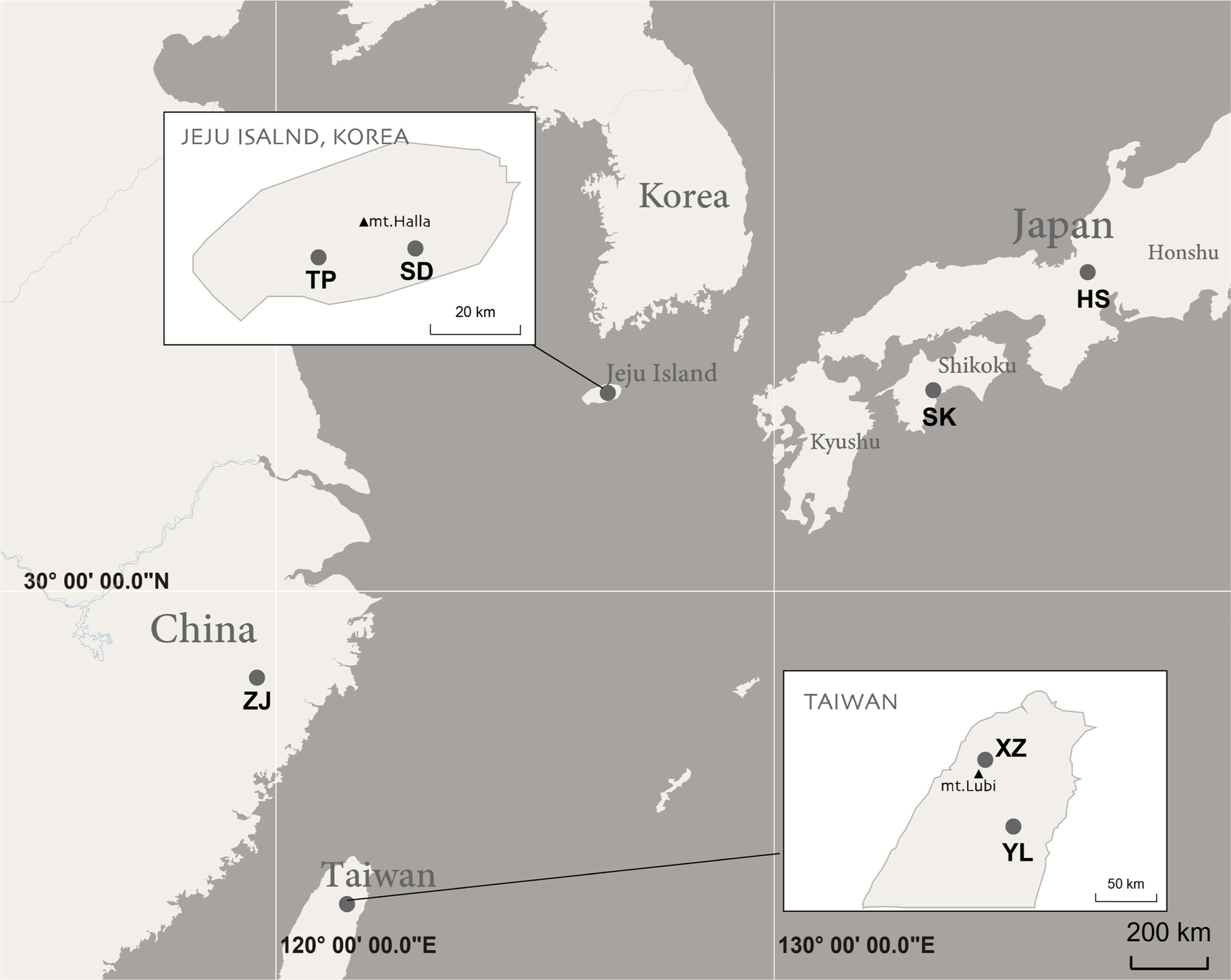
Collecting map of Neottia japonica.

Taxa and accession information included in this study.
DNA extraction, PCR amplification, and sequencing
Extraction of genomic DNA was conducted using DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocols. A matK regions of cpDNA and ITS region of nrDNA were selected to amplify after screening for variable regions. Polymerase Chain Reactions (PCR) was conducted with AccuPower PCR Premix (Bioneer, Daejeon, Korea). Primers used for amplification and sequencing were OMAT1F and trnK2R for matK (Hidayat et al., 2005) and ITS5 and ITS4 for ITS (White et al., 1990). The PCR protocols as follows: 94°C for 5 min; followed by 35 cycles of 94°C for 30 s, 54°C for 1 min, 72°C for 1 min; and a 5 min extension at 72°C. PCR products are purified with the AccuPrep PCR Purification Kit (Bioneer). Complementary strands of the PCR products were directly sequenced with the Big Dye Terminator cycle sequencing reagents (Applied Biosystems, Foster City, CA, USA). Sequencing primers were identical to PCR primers.
Phylogenetic analysis
Forward and reverse sequence fragments were assembled and edited using Geneious Prime version 11.0.6 (Biomatters Ltd., Auckland, New Zealand). Nucleotide sequences of the ITS and cpDNA regions were each aligned with parameter Muscle 3.8.425 (Edgar, 2004) with final adjustment manually. Three taxa of Epipactis, E. papillosa, E. thunbergii, and E. veratrifolia were selected as outgroups according to previous phylogenetic data (Feng et al, 2016; Zhou and Jin, 2018). The data matrices were analyzed initially using maximum parsimony (MP), treating gaps as missing values, and later in separate analyses. Characters were treated as unordered and all character transformations were weighted equally. Heuristic MP searches were replicated 1,000 times with random stepwise addition of taxa, tree-bisection-reconnection (TBR) branch swapping and saving multiple trees. Bootstrap values (Felsenstein, 1985) were calculated from 500 replicate analyses using TBR branch swapping and simple stepwise addition of taxa. We also conducted Bayesian analysis (BI) was performed with MrBayes version 2.2.4 plugged in Geneious Prime version 11.0.6 (Biomatters Ltd., Auckland, New Zealand) to construct the phylogenetic tree. The substitution model was selected as GTR and Monte Carlo Markov Chain analyses were performed for 1,100,000 generations, with sampling done every 500 generations. The results were visualized by FigTree version 1.4.4 (Rambaut, 2016).
Results
The analysis of ITS data included 710 characters from 69 accessions including 36 from N. japonica and three outgroups. A total of 482 (67.0%) were constant and 192 (27.0%) were parsimony informative. The tree length was 433 with a consistency index (CI) of 0.707 and a retention index (RI) of 0.930. Moreover, different branch patterns were observed between the parsimony and Bayesian phylogeny analyses in the clade 1. However, the supporting values were under 50% and therefore could not explain the relationships among the clade 1 in the parsimony phylogram. On the other hand, the posterior probabilities exceeded 70% in the Bayesian phylogram. The placement of N. cordata was different between the parsimony and Bayesian analyses. Specifically, N. cordata was found to be sister to the rest of the Neottia species according to the parsimony analysis, whereas Bayesian phylogeny analysis placed it as a basal group of N. smallii, N. suzukii, and N. japonica. The phylogenetic position of N. cordata from BI is consistent with the cpDNA analyses. The matK gene sequence dataset was aligned to 1,935 bp from 114 accessions including 82 belonging to N. japonica and 2 outgroups. A total of 1,371 (70.6%) characters were constant and 364 (18.8%) characters were parsimony informative. Based on MP analyses, a tree length of 840 with a CI of 0.793 and a RI of 0.9111 were obtained. Bayesian trees were consistent with MP trees. The posterior probabilities from the Bayesian analyses were higher than the bootstrap values from maximum parsimony and the BI topologies, which showed more consensus between cpDNA and nrDNA. Therefore, BI was chosen as the primary tree for discussion in this study (Figs. 3, 4).

Phylogram obtained from Bayesian inference analysis based on internal transcribed spacer of nuclear DNA data. Numbers at nodes show Bayesian posterior probabilities/bootstrap percentages (≥50%), respectively. “-” indicates that the node was not supported in maximum parsimony analysis and */* = 1.0/100.

Phylogram obtained from Bayesian inference analysis based on matK of chloroplast DNA data. Numbers at nodes show Bayesian posterior probabilities/bootstrap percentages (≥50%), respectively. “-” indicates that the node was not supported in maximum parsimony analysis and */* = 1.0/100.
This study included 22 (ITS of nrDNA) and 21 (matK of cpDNA) species of the genus Neottia. Neottia japonica is closely related to N. suzukii and nested within a clade N. smallii and N. cordata (clade 2). The relationship between N. japonica and N. suzukii is different between the nrDNA and cpDNA phylogeny. First, the ITS of nrDNA phylograms indicated that all N. japonica accessions were monophyletic and were sister to N. suzukii. However, matK of cpDNA results indicated that N. japonica from Japan was sister to N. suzukii. The other N. japonica accessions from Korea, Taiwan, and mainland China tended to cluster in a separate clade. Therefore, N. japonica was paraphyletic according to cpDNA phylogeny analyses.
Neottia kiusiana was included for the first time in this phylogeny. This species is mycoheterotrophic (i.e., it lacks leaves and chloroplasts) and is distributed throughout Korea and Japan. According to our phylogenetic analyses, N. kiusiana was sister to N. nidus-avis and a clade containing N. camtschatea, N. himalaica, N. listeroides, and N. acuminata. This clade consists of mycoheterotrophic species with high supporting values both according to ITS and matK phylograms. N. kiusiana and N. nidus-avis nested deeply in Neottia with long branch length.
To determine the relationship between the geographic distribution and phylogenetic relationships of Neottia japonica, individuals collected from two groups in Korea, Japan, and Taiwan and one accession from mainland China were analyzed. Notably, there were inconsistencies between the nrDNA and cpDNA analyses. First, the ITS of the nrDNA phylogeny identified two clades within N. japonica. One clade corresponded with individual samples from Korea, mainland China, and Japan. This clade was subcladed into Korea and mainland China individuals with high supporting value. However, the relationships between Japanese individuals were much less obvious. The other clade consisted of Taiwanese individuals. Second, N. japonica was paraphyletic in the matK of cpDNA phylogeny. The individuals from Korea, Taiwan, and mainland China clustered within the same clade. Moreover, the Taiwan populations were subcladed within the clade, whereas the relationship between Korean populations and mainland China individuals could not be identified. Neither phylogenetic analysis approach could distinguish between populations within the study region.
The auricle’s character was divided into two types. The two type of auricles were classified as “short” when the auricle could not embrace the column (Korea and Taiwan type) and “long” when it embraced the column completely (Japan type). According to ITS phylogeny, the Korea type made a clade with the Japan type. This result shows that the auricle types were not reflected by the nrDNA phylogeny. In matK phylogeny, the Korea type formed a clade with the Taiwan type. However, the Japan type was sister to N. suzukii, which has no auricles. Therefore, ITS and matK phylogeny were unsuitable to identify genetic associations with auricle lengths.
Discussion
Our study conducted a phylogenetic analysis encompassing the largest number of Neottia species to date, and our findings provide a better understanding of the relationships within the genus Neottia compared with previous data (Feng et al., 2016; Zhou and Jin, 2018; Zhu et al., 2019). The taxonomic classification of N. japonica and N. suzukii had been established in previous studies (Zhu et al., 2019), however, this previous phylogenetic analysis included only eight species of Neottia and therefore could not adequately explain inter-specific relationships. Additionally, the phylogenetic position of N. kiusiana, a mycoheterotrophic orchid distributed in Korea and Japan, was elucidated in this study. Phylogenetic analyses included individuals from all regions of Korea, Japan, Taiwan, and mainland China where N. japonica is distributed. Importantly, our findings improve the understanding of the phylogeny and distribution of N. japonica by correlating its geographical distribution, morphological traits, and molecular phylogenetic relationships.
A previous phylogenetic tree identified N. japonica as a close relative of N. suzukii (Zhu et al., 2019). This study included two new N. suzukii individuals from Taiwan (N. suzukii 953, N. suzukii 969) (Table 1) to confirm the phylogenetic relationship between the two taxa. Similar to previous findings, this study also demonstrated that N. japonica and N. suzukii are closely related. Two newly incorporated N. suzukii individuals exhibited the same topology in our study. N. suzukii has similar morphological characters with N. japonica such as narrowly cuneate lip with the deeply 2-lobed apex and column less than 1 mm. Moreover, both of species are flowering mainly in April and show sympatric distribution in Taiwan. Nevertheless, N. japonica and N. suzukii have been previously identified as different species by morphological characters (Su, 2000; Chen et al., 2009). Therefore, it seems clear that these two species have close phylogenetic relationship.
A phylogenetic analysis of combined data of nrDNA and cpDNA presented by Zhou and Jin (2018) identified N. cordata and N. smallii as basal groups within the Neottia genus which are widespread temperate species. In ITS of nrDNA phylogeny, the N. japonica and N. suzukii are sister to N. smallii. N. japonica is relatively widespread in East Asia; however, N. suzukii is only distributed in Taiwan. Previously characterized distribution and molecular phylogenetic relationships were partially consistent with our findings. Alternatively, N. suzukii could be distributed in other regions. N. japonica is paraphyletic in matK of cpDNA phylogeny, and N. japonica from Japan was found to be closely related to N. suzukii from Taiwan although N. japonica and N. suzukii are sympatric in Taiwan. This result suggests that there is genetic association between N. japonica from Japan and N. suzukii from Taiwan. However, it would require further study to confirm this hypothesis by analyzing more populations from both countries with more genetic markers.
The incongruent topologies between nrDNA and cpDNA may be explained by several reasons, such as incomplete lineage sorting, introgression, and ancient hybridization events (Nishimoto et al., 2003; Pelser et al., 2010). It is difficult to identify which process made the incongruence in this study. Moreover, the incongruence is likely invoked by insufficient DNA sequences. It is also possible that hybridization events occur between N. japonica and N. suzukii. N. × veltmanii is hybrid between N. auriculata and N. convallarioides that these three species show sympatric distribution in North America (Catling, 1976). N. japonica and N. suzukii show sympatric distribution in Taiwan, although it is not identified that the distribution of N. suzukii in Japan. Therefore, further distributional and molecular study is needed to confirm the evolutionary history of N. japonica.
All mycoheterotrophic species formed monophyletic clades, which was consistent with previous Neottia phylogenetic analyses (Pridgeon et al., 2005; Xiang et al., 2012; Feng et al., 2016; Zhou and Jin, 2018). Mycoheterotrophic orchids may have independently evolved from autotrophic ancestors within the genus (Zhou and Jin, 2018). This study also demonstrated this evolutionary pattern. Particularly, the reduced plastome sizes and proportions of genes of Neottia acuminata and N. nidus-avis were demonstrated that their evolutionary stages were at rather late stages (Feng et al., 2016). Neottia kiusiana was found to be sister to N. nidus-avis and nested most deeply in the mycoheterotrophic clade. Neottia acuminata, N. nidusavis, and N. kiusiana are known as holomycoheterotrophic orchids (i.e., leafless and achlorophyllous plants), whereas other mycoheterotrophic species in this study are partially green. Therefore, the evolutionary transition of N. kiusiana was inferred at the advanced stage. However, future studies should determine and compare the plastid genome of N. kiusiana with those of other Neottia species.
Based on ITS phylogeny analyses, Korean individuals formed a subclade with one mainland Chinese individual. In matK phylogeny, Korean, Taiwan, and mainland Chinese individuals were claded together. These phylogenetic topologies suggest that the populations of N. japonica in Korea originated from China’s mainland and island. A characterizing regional gene differences could provide useful preliminary data that can later be confirmed with molecular phylogeographic studies (Xie et al., 2012; Sun et al., 2019). On the other hand, the auricle character is not reflected by both of results. Further research is needed on whether there are genetic results that reflect other morphological differences.
Acknowledgements
This study was supported by 2018 creative subject of personal research activation project (개인창의연구과제, project number: 1-2018-0696-001-1) funded by EwhaWomans University.
Notes
Conflict of Interest
The authors declare that there are no conflicts of interest.