The complete chloroplast genome of Erigeron canadensis isolated in Korea (Asteraceae): Insight into the genetic diversity of the invasive species
Article information

Abstract
We have determined the complete chloroplast genome of Erigeron canadensis isolated in Korea. The circular chloroplast genome of E. canadensis is 152,767 bp long and has four subregions: 84,317 bp of large single-copy and 18,446 bp of small single-copy regions are separated by 25,004 bp of inverted repeat regions including 133 genes (88 protein-coding genes, eight rRNAs, and 37 tRNAs). The chloroplast genome isolated in Korea differs from the Chinese isolate by 103 single-nucleotide polymorphisms (SNPs) and 47 insertions and deletion (INDEL) regions, suggesting different invasion sources of E. canadensis in Korea and China. A nucleotide diversity analysis revealed that the trend of the nucleotide diversity of E. canadensis followed that of 11 Erigeron chloroplasts, except for three peaks. The phylogenetic tree showed that our E. canadensis chloroplast is clustered with E. canadensis reported from China. Erigeron canadensis can be a good target when attempting to understand genetic diversity of invasive species.
INTRODUCTION
Horseweed (Erigeron canadensis L.), native to North America, is now widely dispersed throughout the world (Nesom, 1989, 2004). The species was thought to be introduced into Asia in the late 19th century. Since then, it has spread widely to open places, such as roadsides and the margins of farmland and forests as an invasive species, resulting in ecological problems for native plants (Kim, 2005; Kang et al., 2020; Yan et al., 2020). The species has been classified in the genus Conyza based on its morphology, specifically C. canadensis (L.) Cronquist (Cronquist, 1943; Strother, 2006; Susanna and Garcia-Jacas, 2007), generating taxonomic confusion, as two different names have been used for the same species. However, the most recent taxonomic treatments of Asteraceae recognize the species in Erigeron and place Conyza under the synonymy of Erigeron (Chen and Brouillet, 2011; Keil and Nesom, 2012; POWO, 2023). The morphological features of Conyza distinguished from those of Erigeron innclude a reduction of the ligule in the ray floret and a decrease in the number of hermaphroditic disc florets relative to female ray florets. However, these characteristics are also found in some species of Erigeron, supporting the merge of two genera (Noyes, 2000; Strother, 2006). Molecular data have indicated that Conyza is polyphyletic and nested within Erigeron (Noyes, 2000; Brouillet et al., 2009). Erigeron canadensis has been shown to have medicinal potentials given its antifungal (Curini et al., 2003) and anti-platelet activities (Pawlaczyk et al., 2011). As part of the development of molecular marker for the species, we completed the Korean E. canadensis chloroplast genome.
MATERIALS AND METHODS
A plant of the E. canadensis was collected in Gangseo-gu, Seoul, Korea (37.529708N, 126.842867E). A voucher specimen was deposited in the Infoboss Cyber Herbarium (IN, voucher number of IB-30034).
The total DNA was extracted from fresh leaves using a DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). Genome sequencing was performed using NovaSeq6000 at Macrogen Inc., Korea, and de novo assembly was done with Velvet v1.2.10 (Zerbino and Birney, 2008) and GapCloser v1.12 (Zhao et al., 2011). Assembled sequences were confirmed by BWA v0.7.17 (Li, 2013) and SAMtools v1.9 (Li et al., 2009) while separating the complete mitochondrial genome of Uroleucon erigeronense (Park and Lee, 2022). All bioinformatic analyses were conducted in the Genome Information System (GeIS; https://geis.infoboss.co.kr/) as utilized in previous studies (Choi et al., 2021; Kim et al., 2021b; Park et al., 2021a).
Genome annotation was conducted based on another E. canadensis chloroplast (NC_046789) (Zhang et al., 2019) with Geneious R11 v11.0.5 (Biomatters Ltd., Auckland, New Zealand). A circular map of the Korean E. canadensis chloroplast genome was drawn using OGDRAW v1.31 (Greiner et al., 2019).
Single-nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs) were identified from the pair-wise sequence alignment of the two chloroplast genomes of E. canadensis conducted by MAFFT v7.450 (Katoh and Standley, 2013) with the ‘Find variations/SNPs’ function implemented in Geneious R11 v11.0.5 (Biomatters Ltd., Auckland, New Zealand). The INDEL region was defined as continuous INDELs, as in previous studies. All of these analyses were conducted in the GeIS environment, as used in previous studies (Park et al., 2020d; Park et al., 2021c; Yoo et al., 2021; Park and Xi, 2022).
Nucleotide diversity was calculated using the method proposed by Nei and Li (Nei and Li, 1979) based on the multiple-sequence alignment of 11 available Erigeron chloroplast genomes using a Perl script used in previous studies (Kim et al., 2021a; Kim et al., 2021c; Park et al., 2022). To examine the nucleotide diversity throughout the chloroplast genome, we used a sliding-window analysis with a window size of 500 bp and a step size of 200. Genomic coordination of each window was compared to the gene annotation of the chloroplast genome in GeIS.
Maximum-likelihood (ML), neighbor-joining (NJ), and Bayesian inference (BI) phylogenetic trees were constructed based on the multiple-sequence alignment of twenty chloroplast genomes by MAFFT v7.450 (Katoh and Standley, 2013), including that of the outgroup species, Praxelis clematidea (GenBank accession: NC_023833). During the alignment step, chloroplast genomes of Erigeron philadelphicus (GenBank accession: MT579972), Erigeron strigosus (GenBank accession: MT579973), Erigeron multiradiatus (GenBank accession: NC_056169), and two Erigeron annuus types (GenBank accessions: OL350834 and MZ361990) were modified due to the different directions of LSC, SSC, and IRs. The ML and NJ trees were reconstructed in MEGA X (Kumar et al., 2018) with 1,000 and 10,000 bootstrap repeats, respectively. In the ML analysis, a heuristic search was used with nearest-neighbor interchange branch swapping, with the GTR+F+R4 model determined as the best-fit model by jModelTest v2.0.6 (Darriba et al., 2012) and with uniform rates among sites. All other options used the default settings. The posterior probability of each node was estimated by BI using MrBayes v3.2.6 (Huelsenbeck and Ronquist, 2001). The HKY85 model with gamma rates was used as a molecular model. A Markov-chain Monte Carlo algorithm was employed for 1,100,000 generations, sampling trees every 200 generations, with four chains running simultaneously. Trees from the first 100,000 generations were discarded as burn-in.
RESULTS AND DISCUSSION
The E. canadensis chloroplast genome isolated in Korea (GenBank accession: MT806101) is 152,767 bp long (GC ratio is 37.1%) with four subregions: 84,317 bp of large singlecopy (35.0%) regions, 18,446 bp of small single-copy (30.9%) regions, and 25,004 bp of a pair of inverted repeats (43.0%) (Fig. 1). It is slightly longer than NC_046789 (Zhang et al., 2019). It contains 133 genes (88 protein-coding genes, eight rRNAs, and 37 tRNAs); 18 genes (seven protein-coding genes, four rRNAs and seven tRNAs) are duplicated in the IR regions, identical to that of NC_046789.
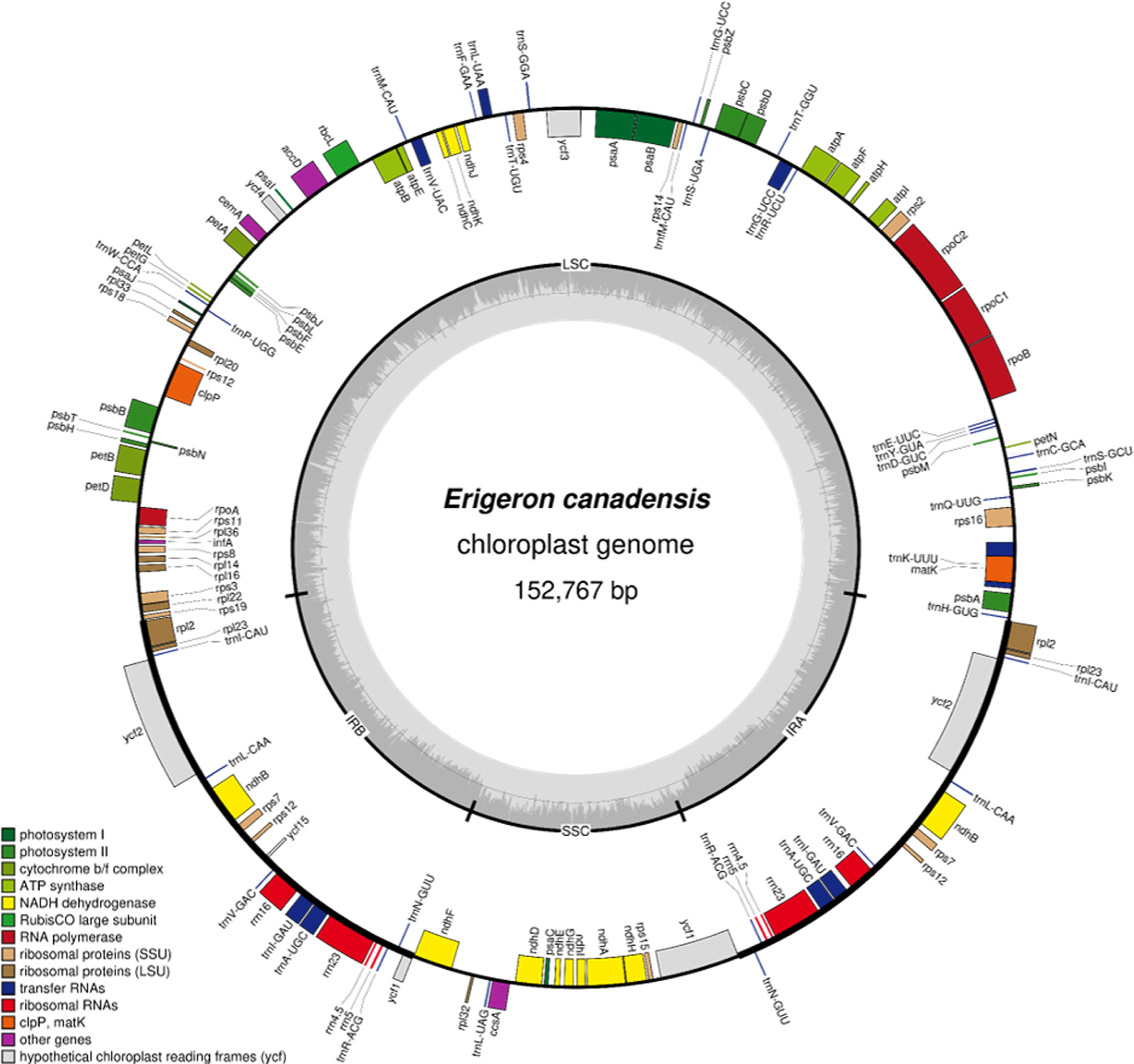
Circular map of the chloroplast genome of Erigeron canadensis isolated in Korea. Genes shown outside are transcribed clockwise and those inside the circle are transcribed in a counter-clockwise arrangement. Genes are color-coded to distinguish different functional groups. The dark grey and light grey plots in the inner circle correspond to the GC content and AT content, respectively.
There were 103 single-nucleotide polymorphisms (SNPs) and 47 insertions and deletion (INDEL) regions (208 bp in total) against the Chinese E. canadensis chloroplast genome (NC_046789). The numbers of intraspecific variations among native populations in closely related species show similar levels. Erigeron breviscapus (Vaniot) Hand.-Mazz., distributed in Western China, displays 70 SNPs and 47 INDEL regions (268 bp in total) between two chloroplast genomes (NC_043882 and MK414770) (Meng et al., 2019; Wang and Lanfear, 2019). Two E. annuus (L.) Pers. chloroplast genomes (OL350834 and MZ361990) collected in China (Zhou et al., 2022) displayed 30 SNPs and 24 INDEL regions (124 bp in total), the lowest number among the four Erigeron species. Thus, the comparison of intraspecific variations suggests that the invasion sources of E. canadensis in the China and Korea cases may be different or that multiple invasions in each country have occurred independently due to the relatively high levels of intraspecific variations. Interestingly, the numbers of intraspecific variations of E. bonariensis L., native to South America, isolated in Western Australia and in the eastern states of Australia (Hereward et al., 2017; Wang et al., 2018a) amount to 105 SNPs and 87 INDEL regions (1,175 bp in length), also exhibiting similar levels of intraspecific variations among invading populations. These patterns suggest that multiple colonization events of pioneer plants, such as E. canadensis and E. bonariensis, may have facilitated the invasiveness (Yang et al., 2012).
The numbers of these intraspecific variations are much higher than those of Artemisia fukudo Makino (Asteraceae; 7 SNPs and 5 INDEL regions (12 bp)) isolated in Korea) (Min et al., 2019) and Suaeda japonica Makino (Chenopodiaceae; 3 SNPs and 3 INDEL regions (3 bp in total) isolated in Korea) (Kim et al., 2020) as well as those of many plant species of which samples have been isolated in Korea and China (Wang et al., 2018b; Jeon et al., 2019; Kim et al., 2019; Park et al., 2019a; Park et al., 2019b; Choi et al., 2020; Park et al., 2020c). However, they are smaller than those of Camellia japonica Wall. (Theaceae; 78 SNPs and 643-bp INDELs) (Park et al., 2019c), Gastrodia elata Blume (Orchidaceae; 457 SNPs and 670-bp INDELs) (Park et al., 2020b), Goodyera schlechtendaliana Rchb. f. (Orchidaceae; 163 to 827 SNPs and 1,060-bp to 1,794-bp INDELs) (Oh et al., 2019a, 2019b), and Selaginella tamariscina (P.Beauv.) Spring (1,213 SNPs and 1,641-bp INDELs) (Park et al., 2020a) isolated in Korea and China. The level of intraspecific variation of the chloroplast genomes can be determined by the rate of molecular evolution, the generation times, and the evolutionary history of each species, and this factor should be investigated with more populations to cover the entire range of variation.
The nucleotide diversity rates between the two chloroplast genomes of E. canadensis and among all 11 Erigeron chloroplast genomes were calculated. The average nucleotide diversity of the two E. canadensis chloroplast genome is 0.0001688, nearly identical to that of Arabidopsis thaliana (L.) Heynh. (Park et al., 2020c), smaller than that of Zoysia japonica Steud. (0.000217) (Lee and Park, 2021), and larger than that of Chenopodium album L. (0.0000625) (Park et al., 2021b), while that of 11 Erigeron chloroplast genomes is 0.002444 (Fig. 2). Eight peaks presenting high nucleotide diversity among the 11 Erigeron chloroplast genomes included three peaks, trnT, psaA-ycf3, and accD-psaI, with no peak of the nucleotide diversity of E. canadensis but with 11 Erigeron chloroplast genomes (Fig. 2A). Among the three peaks, the trnT peak showed that four chloroplast genomes (GenBank accession numbers OL350834, MZ361990, MT579973, and MT579972) lost trnT regions (Fig. 2B) with the accD-psaI peak indicating that MT579972 (E. philadelphicus Willd.) has different sequences in psaI CDS (Fig. 2C). This finding shows that except for these three peaks, the E. canadensis chloroplast genomes follow a trend of nucleotide diversity similar to that found in Erigeron chloroplast genomes.
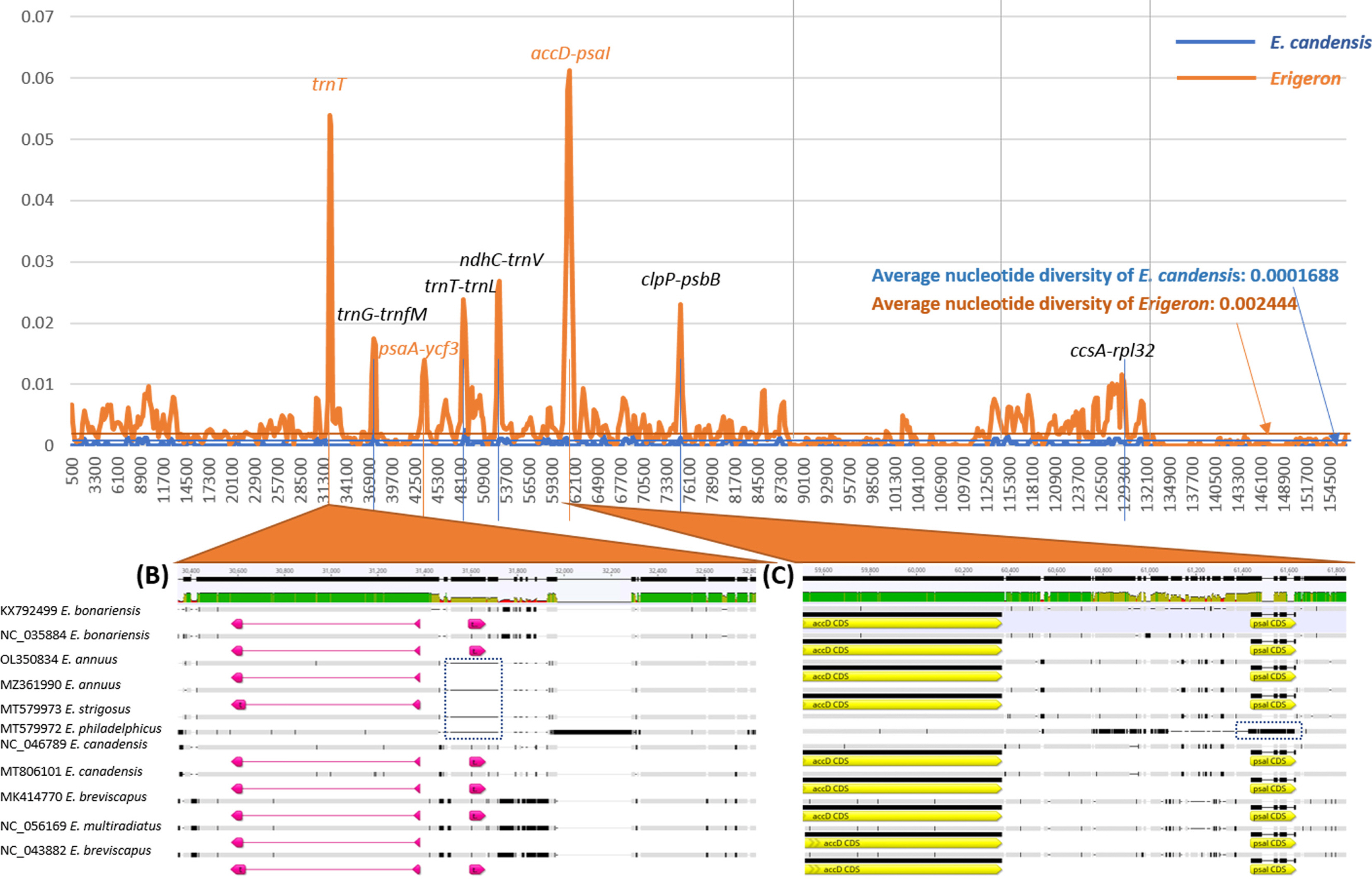
Nucleotide diversity of two E. canadensis and 11 Erigeron chloroplast genomes. (A) displays nucleotide diversities along with the chloroplast genomic coordination of E. canadensis (blue line) and Erigeron (orange line). Labels on the peaks are gene names in the corresponding positions. (B) and (C) show multiple sequence alignments of two regions, the trnT and accD-psaI peaks. The GenBank accessions of 11 Erigeron chloroplast genomes and species names are displayed on the left. Blue dotted boxes indicate different configurations in the alignments.
Twenty Asteraceae chloroplast genomes including one outgroup, Praxelis clematidea (Hieron. ex Kuntze) R. M. King & H. Rob., were used to reconstruct the ML, NJ, and BI phylogenetic trees. All phylogenetic trees indicate that the E. canadensis chloroplast genome assembled in this study is strongly clustered with that of the previously sequenced E. canadensis (Fig. 3). Our phylogenetic analysis of chloroplast genomes also shows that species previously classified in the genus Conyza (i.e., E. bonariensis and E. canadensis) are nested within Erigeron, supporting the broad circumscription of Erigeron (Chen and Brouillet, 2011; Keil and Nesom, 2012; POWO, 2023). The new chloroplast genome data obtained in this study will contribute to a better understand of the genetic diversity of invasive species, which in turn will inform those involved in the management of invasive plants.
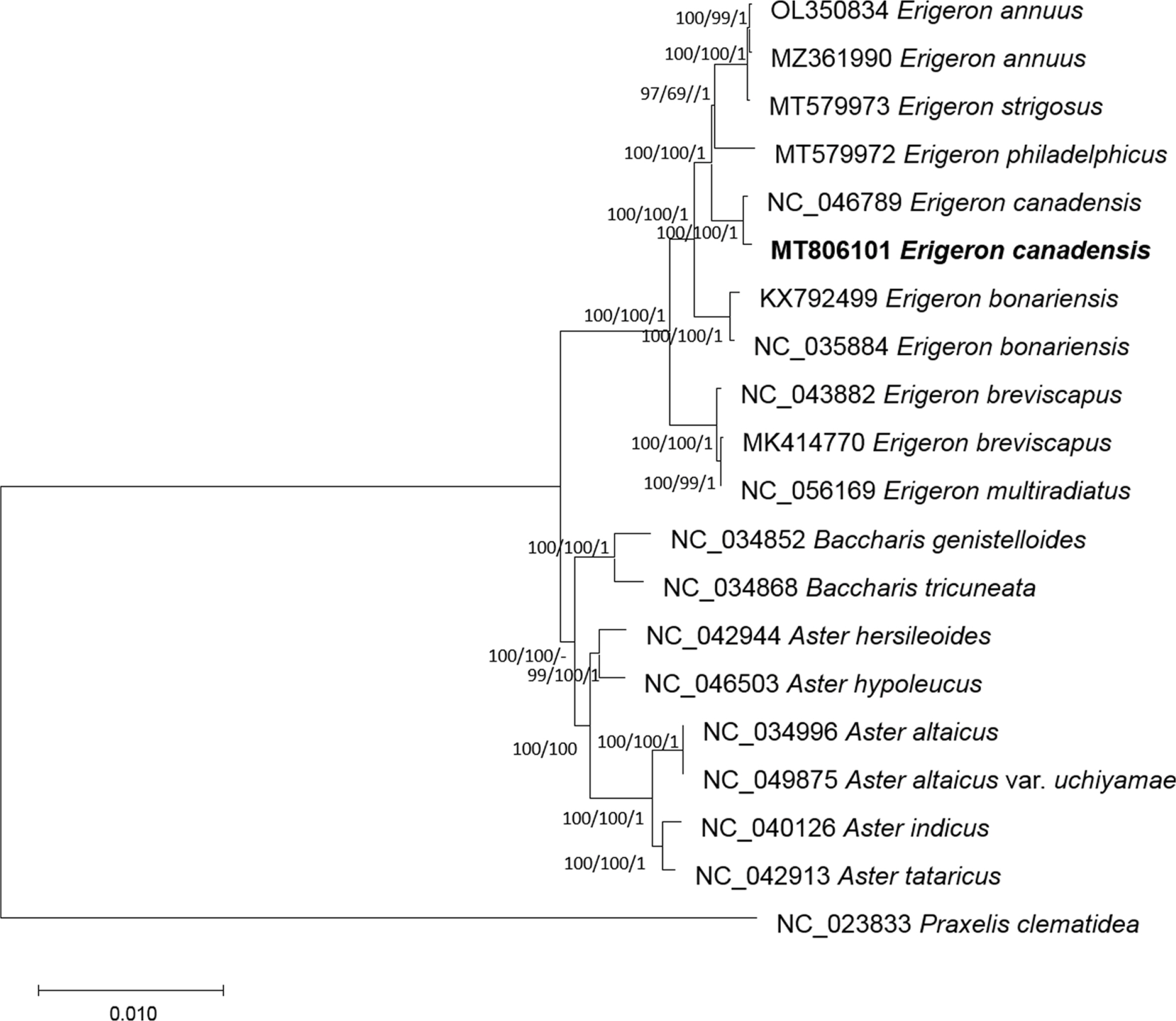
Maximum-likelihood (ML) and Bayesian inference (BI) phylogenetic trees of fifteen chloroplast genomes. Phylogenetic tree was drawn based on the ML tree. The numbers above the branches are the bootstrap support values of the ML and NJ trees and the posterior probabilities from the BI tree.
Acknowledgements
This study was carried out with the support of an InfoBoss Research Grant (IBG-0008) and a research grant from the National Research Foundation of Korea [NRF-2020R1I1A 3068464].
Notes
CONFLICT OF INTEREST
Sang-Hun OH, the Editor-in-Chief of the Korean Journal of Plant Taxonomy, was not involved in the editorial evaluation or decision to publish this article. The authors declare that there are no conflicts of interest.
DATA AVAILABILITY STATEMENT
The chloroplast genome sequence can be accessed via accession number of MT806101 in GenBank of NCBI at https://www.ncbi.nlm.nih.gov. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA688747, SAMN17188058, and SRR13333571, respectively.