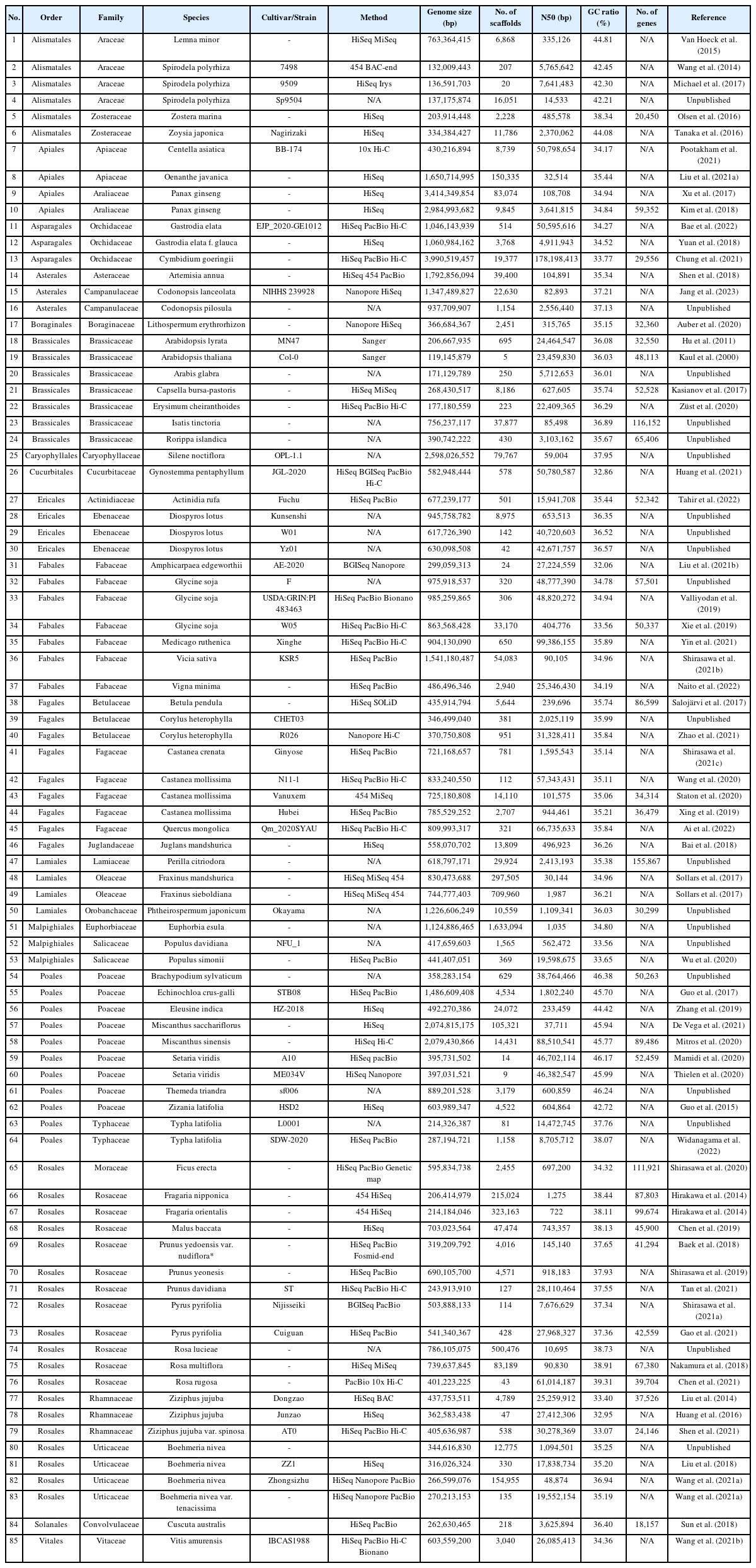
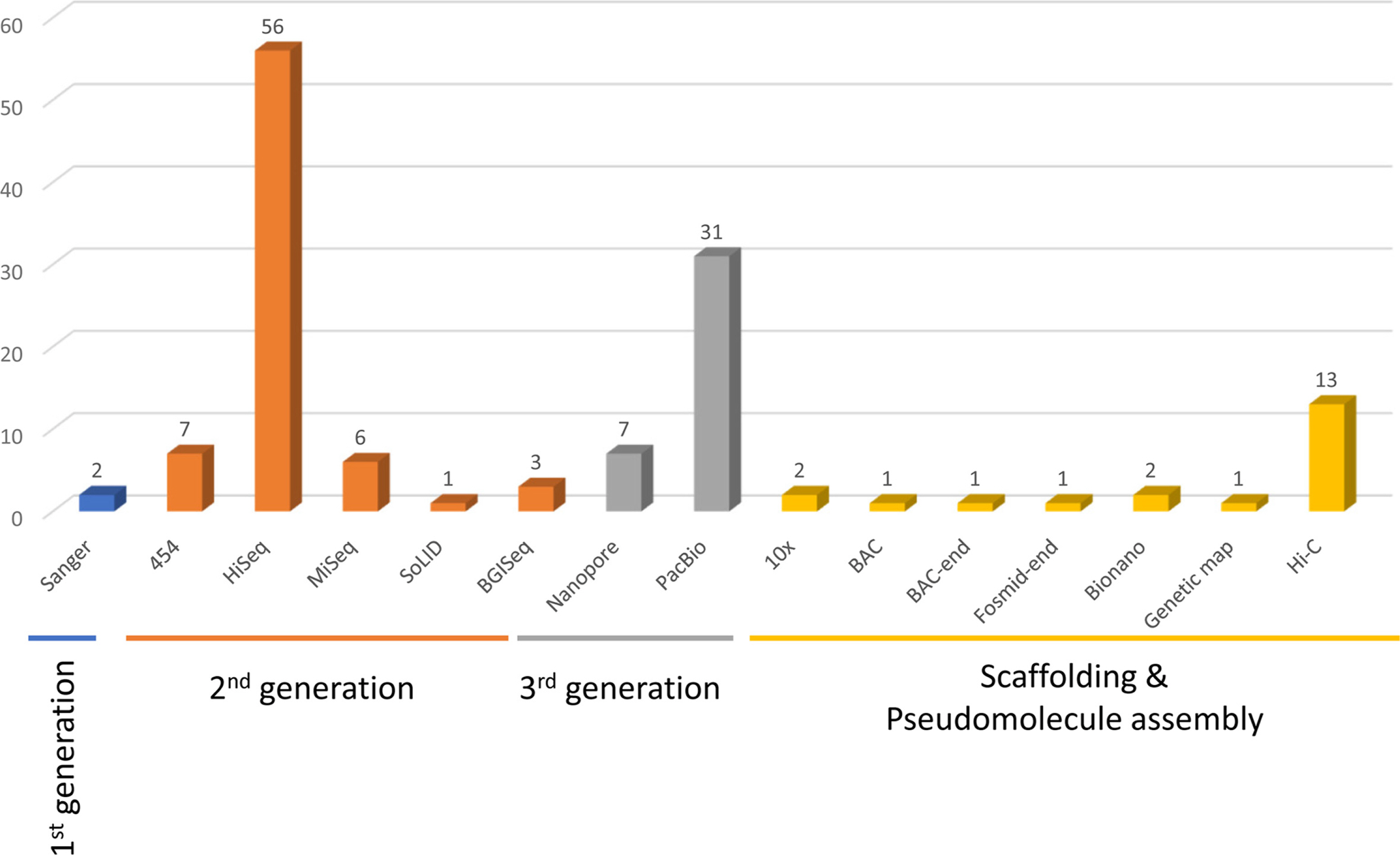
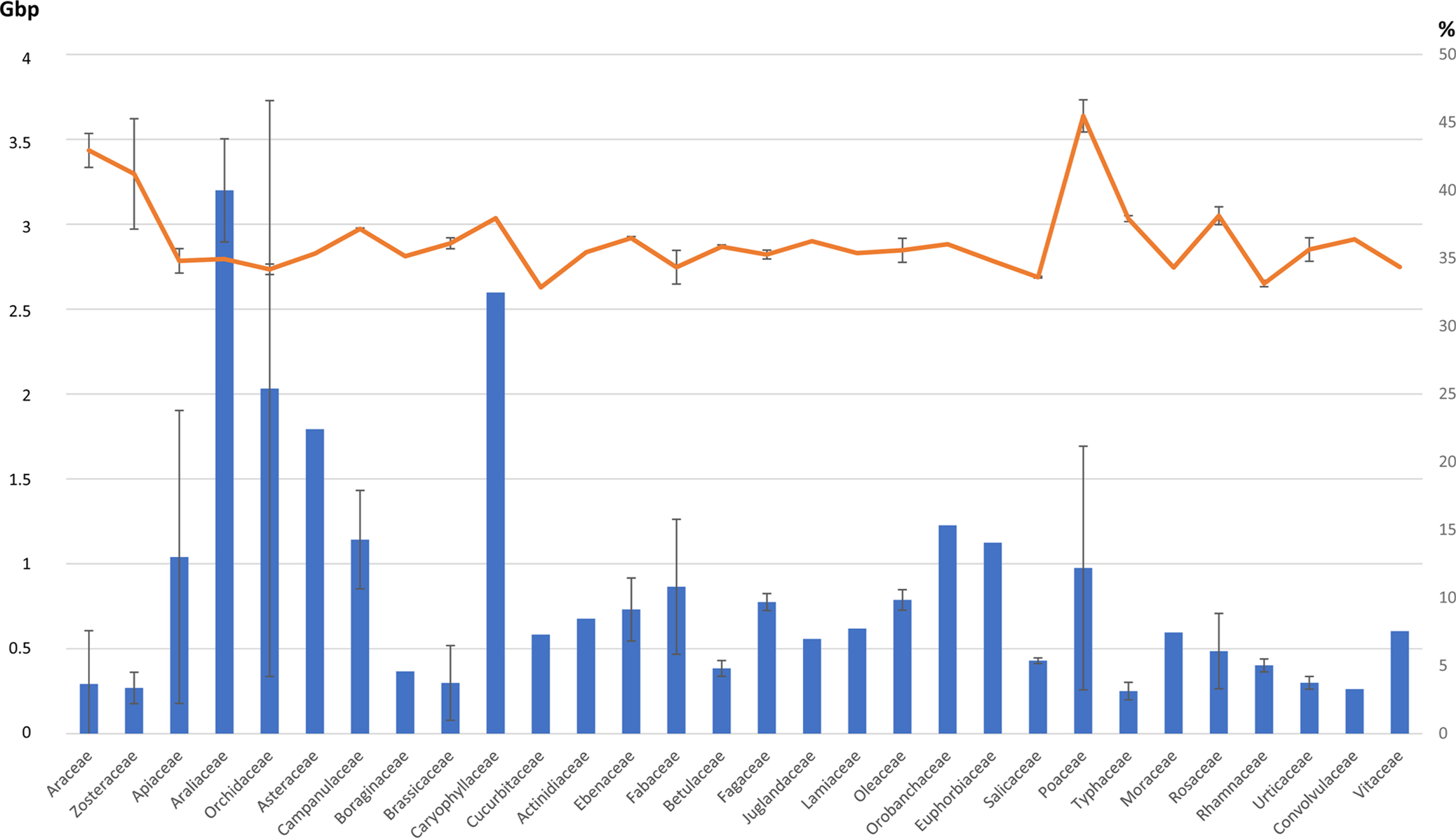
References
Ahmar S, Ballesta P, Ali M, Mora-Poblete F. 2021;Achievements and challenges of genomics-assisted breeding in forest trees: From marker-assisted selection to genome editing. International Journal of Molecular Sciences 22:10583.
Ahmar S, Gill RA, Jung K.-H, Faheem A, Qasim MU, Mubeen M, Zhou W. 2020;Conventional and molecular techniques from simple breeding to speed breeding in crop plants: Recent advances and future outlook. International Journal of Molecular Sciences 21:2590.
Ai W, Liu Y, Mei M, Zhang X, Tan E, Liu H, Han X, Zhan H, Lu X. 2022;A chromosome-scale genome assembly of the Mongolian oak (Quercus mongolica). Molecular Ecology Resources 22:2396–2410.
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. 2002. Molecular Biology of the Cell 4th edth ed. Garland Science. New York: p. 1616.
Amarasinghe S. L, Su S, Dong X, Zappia L, Ritchie M. E, Gouil Q. 2020;Opportunities and challenges in long-read sequencing data analysis. Genome Biology 21:30.
Ansari J. A, Inamdar NN. 2010;The promise of traditional medicines. International Journal of Pharmacology 6:808–812.
Ashelford K, Eriksson M. E, Allen C. M, D'Amore R, Johansson M, Gould P, Kay S, Millar A. J, Hall N, Hall A. 2011;Full genome re-sequencing reveals a novel circadian clock mutation in. Arabidopsis. Genome Biology 12:R28.
Aspinwall M. J, Loik M. E, Resco de Dios V, Tjoelker M. G, Payton P. R, Tissue D. T. 2015;Utilizing intraspecific variation in phenotypic plasticity to bolster agricultural and forest productivity under climate change. Plant, Cell & Environment 38:1752–1764.
Auber R. P, Suttiyut T, McCoy R. M, Ghaste M, Crook J. W, Pendleton A. L, Widhalm J. R, Wisecaver J. H. 2020;Hybrid de novo genome assembly of red gromwell (Lithospermum erythrorhizon) reveals evolutionary insight into shikonin biosynthesis. Horticulture Research 7:82.
Bae E.-K, An C, Kang M.-J, Lee S.-A, Lee S. J, Kim K.-T, Park E.-J. 2022;Chromosome-level genome assembly of the fully mycoheterotrophic orchid Gastrodia elata
. G3 Genes|Genomes|Genetics 12: jkab433.
Baek S, Choi K, Kim G.-B, Yu H.-J, Cho A, Jang H, Kim C, Kim H.-J, Chang K. S, Kim J.-H, Mun J.-H. 2018;Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biology 19:127.
Bai W.-N, Yan P.-C, Zhang B.-W, Woeste K. E, Lin K, Zhang D.- Y. 2018;Demographically idiosyncratic responses to climate change and rapid Pleistocene diversification of the walnut genus Juglans (Juglandaceae) revealed by wholegenome sequences. New Phytologist 217:1726–1736.
Bentley D. R, Balasubramanian S, Swerdlow H. P, Smith G. P, Milton J, Brown C. G, Hall K. P, Evers D. J, Barnes C. L, Bignell H. R, Boutell J. M, Bryant J, Carter R. J, Keira Cheetham R, Cox A. J, Ellis D. J, Flatbush M. R, Gormley N. A, Humphray S. J, Irving L. J, Karbelashvili M. S, Kirk S. M, Li H, Liu X, Maisinger K. S, Murray L. J, Obradovic B, Ost T, Parkinson M. L, Pratt M. R, Rasolonjatovo I. M. J, Reed M. T, Rigatti R, Rodighiero C, Ross M. T, Sabot A, Sankar S. V, Scally A, Schroth G. P, Smith M. E, Smith V. P, Spiridou A, Torrance P. E, Tzonev S. S, Vermaas E. H, Walter K, Wu X, Zhang L, Alam M. D, Anastasi C, Aniebo I. C, Bailey D. M. D, Bancarz I. R, Banerjee S, Barbour S. G, Baybayan P. A, Benoit V. A, Benson K. F, Bevis C, Black P. J, Boodhun A, Brennan J. S, Bridgham J. A, Brown R. C, Brown A. A, Buermann D. H, Bundu A. A, Burrows J. C, Carter N. P, Castillo N, Catenazzi M. C. E, Chang S, Cooley R. N, Crake N. R, Dada O. O, Diakoumakos K. D, Dominguez-Fernandez B, Earnshaw D. J, Egbujor U. C, Elmore D. W, Etchin S. S, Ewan M. R, Fedurco M, Fraser L. J, Fuentes Fajardo K. V, Scott Furey W, George D, Gietzen K. J, Goddard C. P, Golda G. S, Granieri P. A, Green D. E, Gustafson D. L, Hansen N. F, Harnish K, Haudenschild C. D, Heyer NI, Hims M. M, Ho J. T, Horgan A. M, Hoschler K, Hurwitz S, Ivanov D. V, Johnson M. Q, James T, Huw Jones T. A, Kang G.-D, Kerelska T. H, Kersey A. D, Khrebtukova I, Kindwall A. P, Kingsbury Z, Kokko-Gonzales P. I, Kumar A, Laurent M. A, Lawley C. T, Lee S. E, Lee X, Liao A. K, Loch J. A, Lok M, Luo S, Mammen R. M, Martin J. W, McCauley P. G, McNitt P, Mehta P, Moon K. W, Mullens J. W, Newington Taksina, Ning Zemin, Ng Bee Ling, Novo Sonia M, O’Neill Michael J, Osborne M. A, Osnowski A, Ostadan O, Paraschos L. L, Pickering L, Pike A. C, Pike A. C, Chris Pinkard D, Pliskin D. P, Podhasky J, Quijano V. J, Raczy C, Rae V. H, Rawlings S. R, Rodriguez A. C, Roe P. M, Rogers J, Bacigalupo M. C. Rogert, Romanov N, Romieu A, Roth R. K, Rourke N. J, Ruediger S. T, Rusman E, Sanches-Kuiper R. M, Schenker M. R, Seoane J. M, Shaw R. J, Shiver M. K, Short S. W, Sizto NL, Sluis J. P, Smith M. A, Sohna J. E. S, Spence E. J, Stevens K, Sutton N, Szajkowski L, Tregidgo C. L, Turcatti G, vandeVondele S, Verhovsky Y, Virk S. M, Wakelin S, Walcott G. C, Wang J, Worsley G. J, Yan J, Yau L, Zuerlein M, Rogers J, Mullikin J. C, Hurles M. E, McCooke N. J, West J. S, Oaks F. L, Lundberg P. L, Klenerman D, Durbin R, Smith A. J. 2008;Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53–59.
Blankenberg D, Kuster G. V, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. 2010;Galaxy: A web-based genome analysis tool for experimentalists. Current Protocols in Molecular Biology 89:19.10. 11–19.10.21.
Bocklandt S, Hastie A, Cao H. 2019. Bionano genome mapping: high-throughput, ultra-long molecule genome analysis system for precision genome assembly and haploid-resolved structural variation discovery. Single Molecule and Single Cell Sequencing. Advances in Experimental Medicine and Biology 1129In : Suzuki Y, ed. Springer. Singapore: p. 97–118.
Bose S, Banerjee S, Mondal A, Chakraborty U, Pumarol J, Croley C. R, Bishayee A. 2020;Targeting the JAK/STAT signaling pathway using phytocompounds for cancer prevention and therapy. Cells 9:1451.
Buck M, Hamilton C. 2011;The Nagoya Protocol on access to genetic resources and the fair and equitable sharing of benefits arising from their utilization to the Convention on Biological Diversity. Review of European Community & International Environmental Law 20:47–61.
Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, Wang X, Ott F, Müller J, Alonso-Blanco C, Borgwardt K, Schmid K. J, Weigel D. 2011;Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nature Genetics 43:956–963.
Chen F, Su L, Hu S, Xue J.-Y, Liu H, Liu G, Jiang Y, Du J, Qiao Y, Fan Y, Liu H, Yang Q, Lu W, Shao Z.-Q, Zhang J, Zhang L, Chen F, Ceng Z.-M. (Max). 2021;A chromosome- level genome assembly of rugged rose (Rosa rugosa) provides insights into its evolution, ecology, and floral characteristics. Horticulture Research 8:141.
Chen X, Li S, Zhang D, Han M, Jin X, Zhao C, Wang S, Xing L, Ma J, Ji J, An N. 2019;Sequencing of a wild apple (Malus baccata) genome unravels the differences between cultivated and wild apple species regarding disease resistance and cold tolerance. G3: Genes, Genomes, Genetics 9:2051–2060.
Chung G. Y, Jang H.-D, Chang K. S, Choi H. J, Kim Y.-S, Kim H.-J, Son D. C. 2023;A checklist of endemic plants on the Korean Peninsula II. Korean Journal of Plant Taxonomy 53:79–101.
Chung O, Kim J, Bolser D, Kim H.-M, Jun J. H, Choi J.-P, Jang H.- D, Cho Y. S, Bhak J, Kwak M. 2021;A chromosome- scale genome assembly and annotation of the spring orchid (Cymbidium goeringii). Molecular Ecology Resources 22:1168–1177.
De Vega J, Donnison I, Dyer S, Farrar K. 2021;Draft genome assembly of the biofuel grass crop Miscanthus sacchariflorus
. F1000Research 10:29.
Fakhrai-Rad H, Pourmand N, Ronaghi M. 2002;Pyrosequencing: an accurate detection platform for single nucleotide polymorphisms. Human Mutation 19:479–485.
Fleischmann R. D, Adams M. D, White O, Clayton R. A, Kirkness E. F, Kerlavage A. R, Bult C. J, Tomb J. F, Dougherty B. A, Merrick J. M, McKenney K, Sutton G, Fitzhugh C, Fields W, Gocayne J. D, Scott J, Shirley R, Liu L.-L, Glodek A, Kelley J. M, Weidman J. F, Phillips C. A, Spriggs T, Hedblom E, Cotton M. D, Utterback T. R, Hanna M. C, Nguyen D. T, Saudek D. M, Brandon R. C, Fine L. D, Fritchman J. L, Fuhrmann J. L, Geoghagen N. S. M, Gnehm C. L, McDonald L. A, Small K. V, Fraser C, Smith H. O, Craig Venter J. 1995;Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–512.
Fuller C. W, Middendorf L. R, Benner S. A, Church G. M, Harris T, Huang X, Jovanovich S. B, Nelson J. R, Schloss J. A, Schwartz D. C, Vezenov D. V. 2009;The challenges of sequencing by synthesis. Nature Biotechnology 27:1013–1023.
Gan X, Stegle O, Behr J, Steffen J. G, Drewe P, Hildebrand K. L, Lyngsoe R, Schultheiss S. J, Osborne E. J, Sreedharan V. T, Kahles A, Bohnert R, Jean G, Derwent P, Kersey P, Belfield E. J, Harberd N. P, Kemen E, Toomajian C, Kover P. X, Clark R. M, Rätsch G, Mott R. 2011;Multiple reference genomes and transcriptomes for Arabidopsis thaliana
. Nature 477:419–423.
Gao Y, Yang Q, Yan X, Wu X, Yang F, Li J, Wei J, Ni J, Ahmad M, Bai S, Teng Y. 2021;High-quality genome assembly of‘Cuiguan’pear (Pyrus pyrifolia) as a reference genome for identifying regulatory genes and epigenetic modifications responsible for bud dormancy. Horticulture Research 8:197.
Giardine B, Riemer C, Hardison R. C, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent W. J, Nekrutenko A. 2005;Galaxy: a platform for interactive large-scale genome analysis. Genome Research 15:1451–1455.
Goffeau A, Barrell B. G, Bussey H, Davis R. W, Dujon B, Feldmann H, Galibert F, Hoheisel J. D, Jacq C, Johnston M, Louis E. J, Mewes H. W, Murakami Y, Philippsen P, Tettelin H, Oliver S. G. 1996;Life with 6000 genes. Science 274:546–567.
Gulyaev S, Cai X.-J, Guo F.-Y, Kikuchi S, Applequist W. L, Zhang Z.-X, Hörandl E, He L. 2022;The phylogeny of Salix revealed by whole genome re-sequencing suggests different sex-determination systems in major groups of the genus. Annals of Botany 129:485–498.
Guo L, Qiu J, Han Z, Ye Z, Chen C, Liu C, Xin X, Ye C.-Y, Wang Y.-Y, Xie H, Wang Y, Bao J, Tang S, Xu J, Gui Y, Fu F, Wang W, Zhang X, Zhu Q, Guang X, Wang C, Cui H, Cai D, Ge S, Tuskan G. A, Yang X, Qian Q, He S. Y, Wang J, Zhou X.-P, Fan L. 2015;A host plant genome (Zizania latifolia) after a century-long endophyte infection. The Plant Journal 83:600–609.
Guo L, Qiu J, Ye C, Jin G, Mao L, Zhang H, Yang X, Peng Q, Wang Y, Jia L, Lin Z, Li G, Fu F, Liu C, Chen L, Shen E, Wang W, Chu Q, Wu D, Wu S, Xia C, Zhang Y, Zhou X, Wang L, Wu L, Song W, Wang Y, Shu Q, Aoki D, Yumoto E, Yokota T, Miyamoto K, Okada K, Kim D.-S, Cai D, Zhang C, Lou Y, Qian Q, Yamaguchi H, Yamane H, Kong C.-H, Timko M. P, Bai L, Fan L. 2017;
Echinochloa crus-galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nature Communications 8:1031.
Hirakawa H, Shirasawa K, Kosugi S, Tashiro K, Nakayama S, Yamada M, Kohara M, Watanabe A, Kishida Y, Fujishiro T, Tsuruoka H, Minami C, Sasamoto S, Kato M, Nanri K, Komaki A, Yanagi T, Guoxin Q, Maeda F, Ishikawa M, Kuhara S, Sato S, Tabata S, Isobe S. 2014;Dissection of the octoploid strawberry genome by deep sequencing of the genomes of Fragaria species. DNA Research 21:169–181.
Hon T, Mars K, Young G, Tsai Y.-C, Karalius J. W, Landolin J. M, Maurer N, Kudrna D, Hardigan M. A, Steiner C. C, Knapp S. J, Ware D, Shapiro B, Peluso P, Rank D. R. 2020;Highly accurate long-read HiFi sequencing data for five complex genomes. Scientific Data 7:399.
Hu T. T, Pattyn P, Bakker E. G, Cao J, Cheng J.-F, Clark R. M, Fahlgren N, Fawcett J. A, Grimwood J, Gundlach H, Haberer G, Hollister J. D, Ossowski S, Ottilar R. P, Salamov A. A, Schneeberger K, Spannagl M, Wang X, Yang L, Nasrallah M. E, Bergelson J, Carrington J. C, Gaut B. S, Schmutz J, Mayer K. F. X, Van de Peer Y, Grigoriev I. V, Nordborg M, Weigel D, Guo Y.-L. 2011;The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nature Genetics 43:476–481.
Huang D, Ming R, Xu S, Wang J, Yao S, Li L, Huang R, Tan Y. 2021;Chromosome-level genome assembly of Gynostemma pentaphyllum provides insights into gypenoside biosynthesis. DNA Research 28:dsab018.
Huang J, Zhang C, Zhao X, Fei Z, Wan K, Zhang Z, Pang X, Yin X, Bai Y, Sun X, Gao L, Li R, Zhang J, Li X. 2016;The jujube genome provides insights into genome evolution and the domestication of sweetness/acidity taste in fruit trees. PLoS Genetics 12:e1006433.
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas W. J, Wang X, Xie B, Ni P, Ren Y, Zhu H, Li J, Lin K, Jin W, Fei Z, Li G, Staub J, Kilian A, van der Vossen E. A. G, Wu Y, Guo J, He J, Jia Z, Ren Y, Tian G, Lu Y, Ruan J, Qian W, Wang M, Huang Q, Li B, Xuan Z, J. Cao Asan, Wu Z, Zhang J, Cai Q, Bai Y, Zhao B, Han Y, Li Y, Li X, Wang S, Shi Q, Liu S, Cho W. K, Kim J.-Y, Xu Y, Heller-Uszynska K, Miao H, Cheng Z, Zhang S, Wu J, Yang Y, Kang H, Li M, Liang H, Ren X, Shi Z, Wen M, Jian M, Yang H, Zhang G, Yang Z, Chen R, Liu S, Li J, Ma L, Liu H, Zhou Y, Zhao J, Fang X, Li G, Fang L, Li Y, Liu D, Zheng H, Zhang Y, Qin N, Li Z, Yang G, Yang S, Bolund L, Kristiansen K, Zheng H, Li S, Zhang X, Yang H, Wang J, Sun R, Zhang B, Jiang S, Wang J, Du Y, Li S. 2009;The genome of the cucumber, Cucumis sativus L. Nature Genetics 41:1275–1281.
Hutchison C. A III, Chuang R.-Y, Noskov V. N, Assad-Garcia N, Deerinck T. J, Ellisman M. H, Gill J, Kannan K, Karas B. J, Ma L, Pelletier J. F, Qi Z.-Q, Alexander Richter R, Strychalski E. A, Sun L, Suzuki Y, Tsvetanova B, Wise K. S, Smith H. O, Glass J. I, Merryman C, Gibson D. G, Craig Venter J. 2016;Design and synthesis of a minimal bacterial genome. Science 351:aad6253.
International Wheat Genome Sequencing Consortium (IWGSC). 2018;Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191.
Jaillon O, Aury J.-M, Noel B, Policriti A, Clepet C, Cassagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Enrico Pè M, Valle G, Morgante M, Caboche M, Adam-Blondon A.-F, Weissenbach J, Quétier F, Wincker P, ; French-Italian Public Consortium for Grapevine Genome Characterization. 2007;The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467.
Jang W, Kang J.-N, Jo I.-H, Lee S.-M, Park G-H, Kim C-K. 2023;The chromosome-level genome assembly of lance asiabell (Codonopsis lanceolata), a medicinal and vegetable plant of the Campanulaceae family. Frontiers in Genetics 14:1100819.
Jones-Rhoades M. W, Bartel D. P, Bartel B. 2006;MicroR-NAs and their regulatory roles in plants. Annual Review of Plant Biology 57:19–53.
Kang S.-H, Pandey R. P, Lee C.-M, Sim J.-S, Jeong J.-T, Choi B.-S, Jung M, Ginzburg D, Zhao K, Won S. Y, Oh T.-J, Yu Y, Kim N.-H, Lee O. R, Lee T.-H, Bashyal P, Kim T.-S, Lee W.-H, Hawkins C, Kim C.-K, Kim J. S, Ahn B. O, Rhee S. Y, Sohng J. K. 2020;Genome-enabled discovery of anthraquinone biosynthesis in Senna tora
. Nature Communications 11:5875.
Kasianov A. S, Klepikova A. V, Kulakovskiy I. V, Gerasimov E. S, Fedotova A. V, Besedina E. G, Kondrashov A. S, Logacheva M. D, Penin A. A. 2017;High-quality genome assembly of Capsella bursa-pastoris reveals asymmetry of regulatory elements at early stages of polyploid genome evolution. The Plant Journal 91:278–291.
Kim J.-I, Ju Y. S, Park H, Kim S, Lee S, Yi J.-H, Mudge J, Miller N. A, Hong D, Bell C. J, Kim H.-S, Chung I.-S, Lee W.-C, Lee J.-S, Seo S.-H, Yun J.-Y, Woo H. N, Lee H, Suh D, Lee S, Kim H.-J, Yavartanoo M, Kwak M, Zheng Y, Lee M. K, Park H, Kim J. Y, Gokcumen O, Mills R. E, Zaranek A. W, Thakuria J, Wu X, Kim R. W, Huntley J. J, Luo S, Schroth G. P, Wu T. D, Kim H, Yang K.-S, Park W.-Y, Kim H, Church G. M, Lee C, Kingsmore S. F, Seo J.-S. 2009;A highly annotated whole-genome sequence of a Korean individual. Nature 460:1011–1015.
Kim M. Y, Lee S, Van K, Kim T.-H, Jeong S.-C, Choi I.-Y, Kim D.- S, Lee Y.-S, Park D, Ma J, Kim W.-Y, Kim B.-C, Park S, Lee K.-A, Kim D. H, Kim K. H, Shin J. H, Jang Y. E, Kim K. D, Liu W. X, Chaisan T, Kang Y. J, Lee Y.-H, Kim K.- H, Moon J.-K, Schmutz J, Jackson S. A, Bhak J, Lee S.-H. 2010;Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proceedings of the National Academy of Sciences 107:22032–22037.
Kim M, Xi H, Park J. 2021a;Genome-wide comparative analyses of GATA transcription factors among 19 Arabidopsis ecotype genomes: Intraspecific characteristics of GATA transcription factors. PLoS ONE 16:e0252181.
Kim M, Xi H, Park S, Yun Y, Park J. 2021b;Genome-wide comparative analyses of GATA transcription factors among seven Populus genomes. Scientific Reports 11:16578.
Kim N.-H, Jayakodi M, Lee S.-C, Choi B.-S, Jang W, Lee J, Kim H. H, Waminal N. E, Lakshmanan M, van Nguyen B, Lee Y. S, Park H.-S, Koo H. J, Park J. Y, Perumal S, Joh H. J, Lee H, Kim J, Kim I. S, Kim K, Koduru L, Kang K. B, Sung S. H, Yu Y, Park D. S, Choi D, Seo E, Kim S, Kim Y.-C, Hyun D. Y, Park Y.-I, Kim C, Lee T.-H, Kim H. U, Soh M. S, Lee Y, In J. G, Kim H.-S, Kim Y.-M, Yang D.-C, Wing R. A, Lee D.- Y, Paterson A. H, Yang T.-J. 2018;Genome and evolution of the shade-requiring medicinal herb Panax ginseng
. Plant Biotechnology Journal 16:1904–1917.
Kong S, Zhang Y. 2019;Deciphering Hi-C: From 3D genome to function. Cell Biology and Toxicology 35:15–32.
Lee J. E, Neumann M, Duro D. I, Schmid M. 2019;CRISPR-based tools for targeted transcriptional and epigenetic regulation in plants. PLoS ONE 14:e0222778.
Lee J, Park J, Xi H, Park J. 2020;Comprehensive analyses of the complete mitochondrial genome of Figulus binodulus (Coleoptera: Lucanidae). Journal of Insect Science 20:10.
Lewin H. A, Robinson G. E, Kress W. J, Baker W. J, Coddington J, Crandall K. A, Durbin R, Edwards S. V, Forest F, Gilbert M. T. P, Goldstein M. M, Grigoriev I. V, Hackett K. J, Haussler D, Jarvis E. D, Johnson W. E, Patrinos A, Richards S, Castilla-Rubio J. C, van Sluys M.-A, Soltis P. S, Xu X, Yang H, Zhang G. 2018;Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences of the United States of America 115:4325–4333.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie B. R, Sabo P. J, Dorschner M. O, Sandstrom R, Bernstein B, Bender M. A, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny L. A, Lander E. S, Dekker J. 2009;Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293.
Liggett S.B. 2001;Pharmacogenetic applications of the Human Genome project. Nature Medicine 7:281–283.
Liu C, Zeng L, Zhu S, Wu L, Wang Y, Tang S, Wang H, Zheng X, Zhao J, Chen X, Dai Q, Liu T. 2018;Draft genome analysis provides insights into the fiber yield, crude protein biosynthesis, and vegetative growth of domesticated ramie (Boehmeria nivea L. Gaud). DNA Research 25:173–181.
Liu J.-X, Jiang Q, Tao J.-P, Feng K, Li T, Duan A.-Q, Wang H, Xu Z.-S, Liu H, Xiong A-S. 2021a;Integrative genome, transcriptome, microRNA, and degradome analysis of water dropwort (Oenanthe javanica) in response to water stress. Horticulture Research 8:262.
Liu M.-J, Zhao J, Cai Q.-L, Liu G.-C, Wang J.-R, Zhao Z.-H, Liu P, Dai L, Yan G, Wang W.-J, Li X.-S, Chen Y, Sun Y.-D, Liu Z.-G, Lin M.-J, Xiao J, Chen Y.-Y, Li X.-F, Wu B, Ma Y, Jian J.-B, Yang W, Yuan Z, Sun X.-C, Wei Y.-L, Yu L.- L, Zhang C, Liao S.-G, He R.-J, Guang X.-M, Wang Z, Zhang Y.-Y, Luo L.-H. 2014;The complex jujube genome provides insights into fruit tree biology. Nature Communications 5:5315.
Liu Y, Zhang X, Han K, Li R, Xu G, Han Y, Cui F, Fan S, Seim I, Fan G, Li G, Wan S. 2021b;Insights into amphicarpy from the compact genome of the legume Amphicarpaea edgeworthii
. Plant Biotechnology Journal 19:952–965.
Long Q, Rabanal F. A, Meng D, Huber C. D, Farlow A, Platzer A, Zhang Q, Vilhjálmsson B. J, Korte A, Nizhynska V, Voronin V, Korte P, Sedman L, Mandáková T, Lysak M. A, Seren Ü, Hellmann I, Nordborg M. 2013;Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nature Genetics 45:884–890.
Mamidi S, Healey A, Huang P, Grimwood J, Jenkins J, Barry K, Sreedasyam A, Shu S, Lovell J. T, Feldman M, Wu J, Yu Y, Chen C, Johnson J, Sakakibara H, Kiba T, Sakurai T, Tavares R, Nusinow D. A, Baxter I, Schmutz J, Brutnell T. P, Kellogg E. A. 2020;A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nature Biotechnology 38:1203–1210.
Martienssen R, McCombie W. R. 2001;The first plant genome. Cell 105:571–574.
Michael T. P, Bryant D, Gutierrez R, Borisjuk N, Chu P, Zhang H, Xia J, Zhou J, Peng H, El Baidouri M, Hallers B. T, Hastie A. R, Liang T, Acosta K, Gilbert S, McEntee C, Jackson S. A, Mockler T. C, Zhang W, Lam E. 2017;Comprehensive definition of genome features in Spirodela polyrhiza by high-depth physical mapping and short-read DNA sequencing strategies. The Plant Journal 89:617–635.
Miles B. N, Ivanov A. P, Wilson K. A, Doğan F, Japrung D, Edel J. B. 2013;Single molecule sensing with solid-state nanopores: Novel materials, methods, and applications. Chemical Society Reviews 42:15–28.
Mitchell-Olds T. 2001;
Arabidopsis thaliana and its wild relatives: A model system for ecology and evolution. Trends in Ecology and Evolution 16:693–700.
Mitros T, Session A. M, James B. T, Wu G. A, Belaffif M. B, Clark L. V, Shu S, Dong H, Barling A, Holmes J. R, Mattick J. E, Bredeson J. V, Liu S, Farrar K, Głowacka K, Jeżowski S, Barry K, Chae W. B, Juvik J. A, Gifford J, Oladeinde A, Yamada T, Grimwood J, Putnam N. H, Vega J. De, Barth S, Klaas M, Hodkinson T, Li L, Jin X, Peng J, Yu C. Y, Heo K, Yoo J. H, Ghimire B. K, Donnison I. S, Schmutz J, Hudson M. E, Sacks E. J, Moose S. P, Swaminathan K, Rokhsar D. S. 2020;Genome biology of the paleotetraploid perennial biomass crop Miscanthus
. Nature Communications 11:5442.
Moore B. D, Andrew R. L, Külheim C, Foley W. J. 2014;Explaining intraspecific diversity in plant secondary metabolites in an ecological context. New Phytologist 201:733–750.
Naito K, Wakatake T, Shibata T. F, Iseki K, Shigenobu S, Takahashi Y, Ogiso-Tanaka E, Muto C, Teruya K, Shiroma A, Shimoji M, Satou K, Hirano T, Nagano A. J, Tomooka N, Hasebe M, Fukushima K, Sakai H. 2022. Genome sequence of 12
Vigna species as a knowledge base of stress tolerance and resistance. bioRxiv
https://doi.org/10.1101/2022.03.28.486085.
Nakamura N, Hirakawa H, Sato S, Otagaki S, Matsumoto S, Tabata S, Tanaka Y. 2018;Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Research 25:113–121.
Ngo L. T, Okogun JI, Folk WR. 2013;21st century natural product research and drug development and traditional medicines. Natural Product Reports 30:584–592.
Norn S, Permin H, Kruse P. R, Kruse E. 2009;From willow bark to acetylsalicylic acid. Dansk Medicinhistorisk Arbog 37:79–98.
Olsen J. L, Rouzé P, Verhelst B, Lin Y.-C, Bayer T, Collen J, Dattolo E, Paoli E. De, Dittami S, Maumus F, Michel G, Kersting A, Lauritano C, Lohaus R, Töpel M, Tonon T, Vanneste K, Amirebrahimi M, Brakel J, Boström C, Chovatia M, Grimwood J, Jenkins J. W, Jueterbock A, Mraz A, Stam W. T, Tice H, Bornberg-Bauer E, Green P. J, Pearson G. A, Procaccini G, Duarte C. M, Schmutz J, Reusch T. B. H, Van de Peer Y. 2016;The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530:331–335.
Osoegawa K, Mammoser A. G, Wu C, Frengen E, Zeng C, Catanese J. J, de Jong P. J. 2001;A bacterial artificial chromosome library for sequencing the complete human genome. Genome Research 11:483–496.
Ossowski S, Schneeberger K, Clark R. M, Lanz C, Warthmann N, Weigel D. 2008;Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Research 18:2024–2033.
Park J, An J.-H, Kim Y, Kim D, Yang B-G, Kim T. 2020a;Database of National Species List of Korea: The taxonomical systematics platform for managing scientific names of Korean native species. Journal of Species Research 9:233–246.
Park J, Kim M, Xi H, Park S, Yun Y. 2021a. Plant Genome Database Release 2.7: One tera base pairs plant genomes era. In : The 17th KOGO Winter Symposium; 2021 Feb 1–2; Seoul, Korea.
Park J, Lee SH, Kim JH. 2021b;Complete genome sequence of the endosymbiotic bacterium “Candidatus Riesia pediculicola”. Microbiology Resource Announcements 10:e01181–01120.
Park J, Xi H, Han J, Lee J, Kim Y, Lee J.-M, Son J, Ahn J, Jang T, Choi J, Park J. 2020b;Prediction and identification of biochemical pathway of acteoside from whole genome sequences of Abeliophyllum distichum Nakai, cultivar Ok Hwang 1ho. Journal of Convergence for Information Technology 10:76–91.
Park J, Xi H, Kim Y. 2020c;The complete chloroplast genome of Arabidopsis thaliana isolated in Korea (Brassicaceae): An investigation of intraspecific variations of the chloroplast genome of Korean A. thaliana
. International Journal of Genomics 2020:3236461.
Pootakham W, Naktang C, Kongkachana W, Sonthirod C, Yoocha T, Sangsrakru D, Jomchai N, U-thoomporn S, Romyanon K, Toojinda T, Tangphatsornruang S. 2021;
De novo chromosome-level assembly of the Centella asiatica genome. Genomics 113:2221–2228.
Ren L, Guo X, Liu S, Yu T, Guo W, Wang R, Ye S, Lambertini C, Brix H, Eller F. 2020;Intraspecific variation in Phragmites australis: Clinal adaption of functional traits and phenotypic plasticity vary with latitude of origin. Journal of Ecology 108:2531–2543.
Salojärvi J, Smolander O.-P, Nieminen K, Rajaraman S, Safronov O, Safdari P, Lamminmäki A, Immanen J, Lan T, Tanskanen J, Rastas P, Amiryousefi A, Jayaprakash B, Kammonen J. I, Hagqvist R, Eswaran G, Ahonen V. H, Serra J. A, Asiegbu F. O, de Dios Barajas-Lopez J, Blande D, Blokhina O, Blomster T, Broholm S, Brosché M, Cui F, Dardick C, Ehonen S. E, Elomaa P, Escamez S, Fagerstedt K. V, Fujii H, Gauthier A, Gollan PJ, Halimaa P, Heino PI, Himanen K, Hollender C, Kangasjärvi S, Kauppinen L, Kelleher CT, Kontunen-Soppela S, Koskinen JP, Kovalchuk A, Kärenlampi SO, Kärkönen AK, Lim K-J, Leppälä J, Macpherson L, Mikola J, Mouhu K, Mähönen AP, Niinemets Ü, Oksanen E, Overmyer K, Palva ET, Pazouki L, Pennanen V, Puhakainen T, Poczai P, Possen BJHM, Punkkinen M, Rahikainen MM, Rousi M, Ruonala R, van der Schoot C, Shapiguzov A, Sierla M, Sipilä TP, Sutela S, Teeri TH, Tervahauta Arja. I, Vaattovaara A, Vahala J, Vetchinnikova L, Welling A, Wrzaczek M, Xu E, Paulin LG, Schulman AH, Lascoux M, Albert VA, Auvinen P, Helariutta Y, Kangasjärvi J. 2017;Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nature Genetics 49:904–912.
Sanger F, Air G. M, Barrell B. G, Brown NL, Coulson AR, Fiddes JC, Hutchison CA, Slocombe PM, Smith M. 1977;Nucleotide sequence of bacteriophage φX174 DNA. Nature 265:687–695.
Schmitz R. J, Schultz M. D, Urich M. A, Nery J. R, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, Ecker JR. 2013;Patterns of population epigenomic diversity. Nature 495:193–198.
Schnable P. S, Ware D, Fulton R. S, Stein J. C, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves T. A, Minx P, Reily A. D, Courtney L, Kruchowski S. S, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock S. M, Belter E, Du F, Kim K, Abbott R. M, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson S. M, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J, Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K, Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, Kudrna D, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D, Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L, Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S, Faga B, Levy M. J, McMahan L, Van Buren P, Vaughn M. W, Ying K, Yeh C.-T, Emrich S. J, Jia Y, Kalyanaraman A, Hsia A.-P, Barbazuk W. B, Baucom R. S, Brutnell T. P, Carpita N. C, Chaparro C, Chia J.-M, Deragon J.-M, Estill J. C, Fu Y, Jeddeloh J. A, Han Y, Lee H, Li P, Lisch D. R, Liu S, Liu Z, Nagel D. H, McCann M. C, SanMiguel P, Myers A. M, Nettleton D, Nguyen J, Penning B. W, Ponnala L, Schneider K. L, Schwartz D. C, Sharma A, Soderlund C, Springer N. M, Sun Q, Wang H, Waterman M, Westerman R, Wolfgruber T. K, Yang L, Yu Y, Zhang L, Zhou S, Zhu Q, Bennetzen J. L, Dawe R. K, Jiang J, Jiang N, Presting G. G, Wessler S. R, Aluru S, Martienssen R. A, Clifton S. W, McCombie W. R, Wing R. A, Wilson R. K. 2009;The B73 maize genome: Complexity, diversity, and dynamics. Science 326:1112–1115.
Shen L.-Y, Luo H, Wang X-L, Wang X-M, Qiu X-J, Liu H, Zhou S-S, Jia K-H, Nie S, Bao Y-T, Zhang R-G, Yun Q-Z, Chai Y-H, Lu J-Y, Li Y, Zhao S-W, Mao J-F, Jia S-G, Mao Y-M. 2021;Chromosome-scale genome assembly for Chinese sour jujube and insights into its genome evolution and domestication signature. Frontiers in Plant Science 12:773090.
Shen Q, Zhang L, Liao Z, Wang S, Yan T, Shi P, Liu M, Fu X, Pan Q, Wang Y, Lv Z, Lu X, Zhang F, Jiang W, Ma Y, Chen M, Hao X, Li L, Tang Y, Lv G, Zhou Y, Sun X, Brodelius P. E, Rose J. K.C, Tang K. 2018;The genome of Artemisia annua provides insight into the evolution of Asteraceae family and artemisinin biosynthesis. Molecular Plant 11:776–788.
Shirasawa K, Esumi T, Hirakawa H, Tanaka H, Itai A, Ghelfi A, Nagasaki H, Isobe S. 2019;Phased genome sequence of an interspecific hybrid flowering cherry,‘Somei- Yoshino’(Cerasus× yedoensis). DNA Research 26:379–389.
Shirasawa K, Itai A, Isobe S. 2021a;Chromosome-scale genome assembly of Japanese pear (Pyrus pyrifolia) variety ‘Nijisseiki’. DNA Research 28:dsab001.
Shirasawa K, Kosugi S, Sasaki K, Ghelfi A, Okazaki K, Toyoda A, Hirakawa H, Isobe S. 2021b;Genome features of common vetch (Vicia sativa) in natural habitats. Plant Direct 5:e352.
Shirasawa K, Nishio S, Terakami S, Botta R, Marinoni D. T, Isobe S. 2021c;Chromosome-level genome assembly of Japanese chestnut (Castanea crenata Sieb. et Zucc.) reveals conserved chromosomal segments in woody rosids. DNA Research 28:dsab016.
Shirasawa K, Yakushiji H, Nishimura R, Morita T, Jikumaru S, Ikegami H, Toyoda A, Hirakawa H, Isobe S. 2020;The Ficus erecta genome aids Ceratocystis canker resistance breeding in common fig (F. carica). The Plant Journal 102:1313–1322.
Slavov G. T, DiFazio S. P, Martin J, Schackwitz W, Muchero W, Rodgers-Melnick E, Lipphardt M. F, Pennacchio C. P, Hellsten U, Pennacchio L. A, Gunter L. E, Ranjan P, Vining K, Pomraning K. R, Wilhelm L. J, Pellegrini M, Mockler T. C, Freitag M, Geraldes A, El-Kassaby Y. A, Mansfield S. D, Cronk Q. C. B, Douglas C. J, Strauss S. H, Rokhsar D, Tuskan G. A. 2012;Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa
. New Phytologist 196:713–725.
Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, Pacini E, Tichý L, Grulich V, Rotreklová O. 2014;Ecological and evolutionary significance of genomic GC content diversity in monocots. Proceedings of the National Academy of Sciences of the United States of America 111:E4096–E4102.
Smith A. M, Heisler L. E, Onge R. P. St, Farias-Hesson E, Wallace I. M, Bodeau J, Harris A. N, Perry K. M, Giaever G, Pourmand N, Nislow C. 2010;Highly-multiplexed barcode sequencing: An efficient method for parallel analysis of pooled samples. Nucleic Acids Research 38:e142.
Sollars E. S. A, Harper A. L, Kelly L. J, Sambles C. M, Ramirez-Gonzalez R. H, Swarbreck D, Kaithakottil G, Cooper E. D, Uauy C, Havlickova L, Worswick G, Studholme D. J, Zohren J, Salmon D. L, Clavijo B. J, Li Y, He Z, Fellgett A, McKinney L. V, Nielsen L. R, Douglas G. C, Kjær E. D, Downie J. A, Boshier D, Lee S, Clark J, Grant M, Bancroft I, Caccamo M, Buggs R. J. A. 2017;Genome sequence and genetic diversity of European ash trees. Nature 541:212–216.
Song X, Li Y, Cao X, Qi Y. 2019;MicroRNAs and their regulatory roles in plant–environment interactions. Annual Review of Plant Biology 70:489–525.
Sprink T, Wilhelm R, Hartung F. 2022;Genome editing around the globe: An update on policies and perceptions. Plant Physiology 190:1579–1587.
Staton M, Addo-Quaye C, Cannon N, Yu J, Zhebentyayeva T, Huff M, Islam-Faridi N, Fan S, Georgi LL, Nelson CD, Bellis E, Fitzsimmons S, Henry N, Drautz-Moses D, Noorai RE, Ficklin S, Saski C, Mandal M, Wagner TK, Zembower N, Bodénès C, Holliday J, Westbrook J, Lasky J, Hebard FV, Schuster SC, Abbott A. G, Carlson J. E. 2020;A reference genome assembly and adaptive trait analysis of Castanea mollissima ‘Vanuxem,’ a source of resistance to chestnut blight in restoration breeding. Tree Genetics and Genomes 16:57.
Sun G, Xu Y, Liu H, Sun T, Zhang J, Hettenhausen C, Shen G, Qi J, Qin Y, Li J, Wang L, Chang W, Guo Z, Baldwin I. T, Wu J. 2018;Large-scale gene losses underlie the genome evolution of parasitic plant Cuscuta australis
. Nature Communications 9:2683.
Tahir J, Crowhurst R, Deroles S, Hilario E, Deng C, Schaffer R, Lievre L. Le, Brendolise C, Chagne D, Gardiner S. E, Knaebel M, Catanach A, McCallum J, Datson P, Thomson S, Brownfield L. R, Nardozza S, Pilkington S. M. 2022;First chromosome-scale assembly and deep floral-bud transcriptome of a male kiwifruit. Frontiers in Genetics 13:852161.
Tan Q, Li S, Zhang Y, Chen M, Wen B, Jiang S, Chen X, Fu X, Li D, Wu H, Wang Y, Xiao W, Li L. 2021;Chromosome- level genome assemblies of five Prunus species and genome-wide association studies for key agronomic traits in peach. Horticulture Research 8:213.
Tanaka H, Hirakawa H, Kosugi S, Nakayama S, Ono A, Watanabe A, Hashiguchi M, Gondo T, Ishigaki G, Muguerza M, Shimizu K, Sawamura N, Inoue T, Shigeki Y, Ohno N, Tabata S, Akashi R, Sato S. 2016;Sequencing and comparative analyses of the genomes of zoysiagrasses. DNA Research 23:171–180.
The 1001 Genomes Consortium. 2016;1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana
. Cell 166:481–491.
The 3,000 Rice Genomes Project. 2014;The 3,000 rice genomes project. GigaScience 3:7.
The Arabidopsis Genome Initiative. 2000;Analysis of the genome sequence of the flowering plant Arabidopsis thaliana
. Nature 408:796–815.
Thielen P. M, Pendleton A. L, Player R. A, Bowden K. V, Lawton T. J, Wisecaver J. H. 2020;Reference genome for the highly transformable Setaria viridis ME034V. G3: Genes, Genomes, Genetics 10:3467–3478.
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen G-L, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Déjardin A, Depamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J, Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, Henrissat B, Holligan D, Holt R, Huang W, Islam-Faridi N, Jones S, Jones-Rhoades M, Jorgensen R, Joshi C, Kangasjärvi J, Karlsson J, Kelleher C, Kirkpatrick R, Kirst M, Kohler A, Kalluri U, Larimer F, Leebens-Mack J, Leplé J-C, Locascio P, Lou Y, Lucas S, Martin F, Montanini B, Napoli C, Nelson D. R, Nelson C, Nieminen K, Nilsson O, Pereda V, Peter G, Philippe R, Pilate G, Poliakov A, Razumovskaya J, Richardson P, Rinaldi C, Ritland K, Rouzé P, Ryaboy D, Schmutz J, Schrader J, Segerman B, Shin H, Siddiqui A, Sterky F, Terry A, Tsai C-J, Uberbacher E, Unneberg P, Vahala J, Wall K, Wessler S, Yang G, Yin T, Douglas C, Marra M, Sandberg G, Van de Peer Y, Rokhsar D. 2006;The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604.
Valliyodan B, Cannon S. B, Bayer P. E, Shu S, Brown A. V, Ren L, Jenkins J, Chung C. Y.-L, Chan T.-F, Daum C. G, Plott C, Hastie A, Baruch K, Barry K. W, Huang W, Patil G, Varshney R. K, Hu H, Batley J, Yuan Y, Song Q, Stupar R. M, Goodstein D. M, Stacey G, Lam H.-M, Jackson S. A, Schmutz J, Grimwood J, Edwards D, Nguyen H. T. 2019;Construction and comparison of three reference-quality genome assemblies for soybean. The Plant Journal 100:1066–1082.
Van Hoeck A, Horemans N, Monsieurs P, Cao H. X, Vandenhove H, Blust R. 2015;The first draft genome of the aquatic model plant Lemna minor opens the route for future stress physiology research and biotechnological applications. Biotechnology for Biofuels 8:188.
Venter J. C, Adams M. D, Myers E. W, Li P. W, Mural R. J, Sutton G. G, Smith H. O, Yandell M, Evans C. A, Holt R. A, Gocayne J. D, Amanatides P, Ballew R. M, Huson D. H, Wortman J. R, Zhang Q, Kodira C. D, Zheng X. H, Chen L, Skupski M, Subramanian G, Thomas P. D, Zhang J, Gabor Miklos G. L, Nelson C, Broder S, Clark A. G, Nadeau J, McKusick V. A, Zinder N, Levine A. J, Roberts R. J, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannen-halli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian A. E, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman T. J, Higgins M. E, Ji R. R, Ke Z, Ketchum K. A, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov G. V, Milshina N, Moore H. M, Naik A. K, Narayan V. A, Neelam B, Nusskern D, Rusch D. B, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng M. L, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers Y. H, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint N. N, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril J. F, Guigó R, Campbell M. J, Sjolander K. V, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang Y. H, Coyne M, Dahlke C, Deslattes Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. 2001;The sequence of the human genome. Science 291:1304–1351.
Wang J, Tian S, Sun X, Cheng X, Duan N, Tao J, Shen G. 2020;Construction of pseudomolecules for the Chinese chestnut (Castanea mollissima) genome. G3: Genes, Genomes, Genetics 10:3565–3574.
Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, Guo Y, Feng B, Li H, Lu Y, Fang X, Liang H, Du Z, Li D, Zhao Y, Hu Y, Yang Z, Zheng H, Hellmann I, Inouye M, Pool J, Yi X, Zhao J, Duan J, Zhou Y, Qin J, Ma L, Li G, Yang Z, Zhang G, Yang B, Yu C, Liang F, Li W, Li S, Li D, Ni P, Ruan J, Li Q, Zhu H, Liu D, Lu Z, Li S, Guo G, Zhang J, Ye J, Fang L, Hao Q, Chen Q, Liang Y, Su Y, San A, Ping C, Yang S, Chen F, Li L, Zhou K, Zheng H, Ren Y, Yang L, Gao Y, Yang G, Li Z, Feng X, Kristiansen K, Wong G. K.-S, Nielsen R, Durbin R, Bolund L, Zhang X, Li S, Yang H, Wang J. 2008;The diploid genome sequence of an Asian individual. Nature 456:60–65.
Wang W, Haberer G, Gundlach H, Gläßer C, Nussbaumer T, Luo M, Lomsadze A, Borodovsky M, Kerstetter R, Shanklin J, Byrant D. W, Mockler T. C, Appenroth K. J, Grimwood J, Jenkins J, Chow J, Choi C, Adam C, Cao X.-H, Fuchs J, Schubert I, Rokhsar D, Schmutz J, Michael T. P, Mayer K. F. X, Messing J. 2014;The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nature Communications 5:3311.
Wang Y, Li F, He Q, Bao Z, Zeng Z, An D, Zhang T, Yan L, Wang H, Zhu S, Liu T. 2021a;Genomic analyses provide comprehensive insights into the domestication of bast fiber crop ramie (Boehmeria nivea). The Plant Journal 107:787–800.
Wang Y, Xin H, Fan P, Zhang J, Liu Y, Dong Y, Wang Z, Yang Y, Zhang Q, Ming R, Zhong G-Y, Li S, Liang Z. 2021b;The genome of Shanputao (Vitis amurensis) provides a new insight into cold tolerance of grapevine. The Plant Journal 105:1495–1506.
Weber J. L, Myers E. W. 1997;Human whole-genome shotgun sequencing. Genome Research 7:401–409.
Weisenfeld NI, Kumar V, Shah P, Church D. M, Jaffe D. B. 2017;Direct determination of diploid genome sequences. Genome Research 27:757–767.
Widanagama S. D, Freeland J. R, Xu X, Shafer A. B. A. 2022;Genome assembly, annotation, and comparative analysis of the cattail Typha latifolia
. G3: Genes, Genomes, Genetics 12:jkab401.
Williams L. J. S, Tabbaa DG, Li N, Berlin AM, Shea TP, MacCallum I, Lawrence MS, Drier Y, Getz G, Young SK, Jaffe DB, Nusbaum C, Gnirke A. 2012;Paired-end sequencing of Fosmid libraries by Illumina. Genome Research 22:2241–2249.
Wu H, Yao D, Chen Y, Yang W, Zhao W, Gao H, Tong C. 2020;
De novo genome assembly of Populus simonii further supports that Populus simonii and Populus trichocarpa belong to different sections. G3: Genes, Genomes, Genetics 10:455–466.
Xie M, Chung C. Y.-L, Li M.-W, Wong F.-L, Wang X, Liu A, Wang Z, Leung A. K.-Y, Wong T.-H, Tong S.-W, Xiao Z, Fan K, Ng M.-S, Qi X, Yang L, Deng T, He L, Chen L, Fu A, Ding Q, He J, Chung G, Isobe S, Tanabata T, Valliyodan B, Nguyen H. T, Cannon S. B, Foyer C. H, Chan T.-F, Lam H.-M. 2019;A reference-grade wild soybean genome. Nature Communications 10:1216.
Xing Y, Liu Y, Zhang Q, Nie X, Sun Y, Zhang Z, Li H, Fang K, Wang G, Huang H, isseling T, Cao Q, Qin L. 2019;Hybrid de novo genome assembly of Chinese chestnut (Castanea mollissima). GigaScience 8:giz112.
Xu J, Chu Y, Liao B, Xiao S, Yin Q, Bai R, Su H, Dong L, Li X, Qian J, Zhang J, Zhang Y, Zhang X, Wu M, Zhang J, Li G, Zhang L, Chang Z, Zhang Y, Jia Z, Liu Z, Afreh D, Nahurira R, Zhang L, Cheng R, Zhu Y, Zhu G, Rao W, Zhou C, Qiao L, Huang Z, Cheng Y.-C, Chen S. 2017;
Panax ginseng genome examination for ginsenoside biosynthesis. GigaScience 6:gix093.
Yin M, Zhang S, Du X, Mateo R. G, Guo W, Li A, Wang Z, Wu S, Chen J, Liu J, Ren G. 2021;Genomic analysis of Medicago ruthenica provides insights into its tolerance to abiotic stress and demographic history. Molecular Ecology Resources 21:1641–1657.
Yuan Y, Jin X, Liu J, Zhao X, Zhou J, Wang X, Wang D, Lai C, Xu W, Huang J, Zha L, Liu D, Ma X, Wang L, Zhou M, Jiang Z, Meng H, Peng H, Liang Y, Li R, Jiang C, Zhao Y, Nan T, Jin Y, Zhan Z, Yang J, Jiang W, Huang L. 2018;The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nature Communications 9:1615.
Zhang H, Hall N, Goertzen LR, Bi B, Chen CY, Peatman E, Lowe EK, Patel J, McElroy J. S. 2019;Development of a goosegrass (Eleusine indica) draft genome and application to weed science research. Pest Management Science 75:2776–2784.
Zhang H.-B, Wu C. 2001;BAC as tools for genome sequencing. Plant Physiology and Biochemistry 39:195–209.
Zhao T, Ma W, Yang Z, Liang L, Chen X, Wang G, Ma Q, Wang L. 2021;A chromosome-level reference genome of the hazelnut, Corylus heterophylla Fisch. GigaScience 10:giab027.
Zimin A, Stevens KA, Crepeau MW, Holtz-Morris A, Koriabine M, Marçais G, Puiu D, Roberts M, Wegrzyn JL, de Jong PJ, Neale DB, Salzberg SL, Yorke J. A, Langley C. H. 2014;Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics 196:875–890.
Zou Y.-P, Hou X.-H, Wu Q, Chen J.-F, Li Z.-W, Han T.-S, Niu X.- M, Yang L, Xu Y.-C, Zhang J, Zhang F.-M, Tan D, Tian Z, Gu H, Guo Y.-L. 2017;Adaptation of Arabidopsis thaliana to the Yangtze River basin. Genome Biology 18:239.
Züst T, Strickler SR, Powell AF, Mabry ME, An H, Mirzaei M, York T, Holland CK, Kumar P, Erb M, Petschenka G, Gómez J-M, Perfectti F, Müller C, Pires JC, Mueller L. A, Jander G. 2020;Independent evolution of ancestral and novel defenses in a genus of toxic plants (Erysimum, Brassicaceae). Elife 9:e51712.