PARK, YUN, XI, KWON, and SON: Current status of whole-genome sequences of Korean angiosperms
Abstract
Owing to the rapid development of sequencing technologies, more than 1,000 plant genomes have been sequenced and released. Among them, 69 Korean plant taxa (85 genome sequences) contain at least one whole-genome sequence despite the fact that some samples were not collected in Korea. The sequencing-by-synthesis method (next-generation sequencing) and the PacBio (third-generation sequencing) method were the most commonly used in studies appearing in 65 publications. Several scaffolding methods, such as the Hi-C and 10x types, have also been used for pseudo-chromosomal assembly. The most abundant families among the 69 taxa are Rosaceae (10 taxa), Brassicaceae (7 taxa), Fabaceae (7 taxa), and Poaceae (7 taxa). Due to the rapid release of plant genomes, it is necessary to assemble the current understanding of Korean plant species not only to understand their whole genomes as our own plant resources but also to establish new tools for utilizing plant resources efficiently with various analysis pipelines, including AI-based engines.
Keywords: Korean angiosperm species, plant resource utilization, scaffolding technology, sequencing technology, taxonomical distribution, Whole-genome sequence
INTRODUCTION
A genome sequence consisting of the complete list of nucleotides contains all of the genetic information of an organism ( Alberts et al., 2002). Once the genome is deciphered properly, theoretically we may find all information pertaining to an organism, thus providing strong motivation for several early genome projects ( Liggett, 2001), including those focusing on Arabidopsis thaliana L. ( Martienssen and McCombie 2001; Mitchell-Olds, 2001). In 2000, the first plant whole-genome sequence, five chromosomes of A. thaliana L., was fully assembled and published ( The Arabidopsis Genome Initiative, 2000), marking a relatively late start in comparison to those of bacteriophages ( Sanger et al., 1977), bacteria ( Fleischmann et al., 1995), and fungi ( Goffeau et al., 1996). At that time, the A. thaliana genome had 25,498 genes ( The Arabidopsis Genome Initiative, 2000), considered to be the most important element to understanding plants ( Hutchison et al., 2016). Over time, researchers realized that additional elements, such as non-coding RNAs, also played important roles ( Jones-Rhoades et al., 2006; Song et al., 2019) as other aspects by which we could understand plant whole-genome sequences. Whole-genome sequences have been assembled from small pieces of DNA from organisms due to two major limitations of sequencing: (1) the start position of sequencing cannot be determined in a whole-genome project, and (2) the length of sequences we can obtain is limited, e.g., 500 bp to 1,000 bp when using the Sanger sequencing method. To decipher whole-genome sequences with sufficiently long lengths (e.g., A. thaliana is 119 Mbp ( The Arabidopsis Genome Initiative, 2000) and Zea mays L. is 2.3 Gbp ( Schnable et al., 2009)), an approach known as the whole-shotgun strategy was adopted after a method based on bacterial artificial chromosome (BAC) sequences ( Zhang and Wu, 2001) given the large-scale computing power for genome assembly ( Weber and Myers, 1997). After the commercialization of next-generation sequencing (NGS) technologies, which have critically reduced sequencing costs, to promote whole-genome sequencing projects, many plant genome sequences were released. The National Center for Biotechnology Information (NCBI) has served as a data depository for many plant genome sequences (e.g., Cymbidium goeringii (Rchb.f.) Rchb.f. ( Chung et al., 2021)). A centralized database in which to archive plant genome sequences is useful to provide access for comparative genomic analyses. The Plant Genome Database ( https://www.plantgenome.info/) is such an example of a centralized database for plant whole-genome sequences ( Park et al., 2021a), containing 3,509 genomes originating from 1,431 species (Release 3.0 to be available in March 2023). These genomic resources are important to conduct further various comparative genomic analyses to understand the genomes. The accumulation of genome data for eukaryotes on a global scale has already begun ( Lewin et al., 2018). Synthesizing the current status of whole-genome data on a local scale is necessary to set the stage for future research to ascertain the biodiversity and evolution of Korean plants and to discern applications of genomic resources ultimately to conserve endangered species. In this review, we examine the historical background of genome sequencing for plants, emphasizing the development of various sequencing technologies and the current status of whole genomes for angiosperms in Korea.
HISTORY OF SEQUENCING TECHNOLOGIES UTILIZED FOR DECIPHERING PLANT GENOMES
Sequencing technologies have been improved based on the requirements of whole-genome projects. First, it is now possible to increase the number of reads when conducting a whole-genome assembly given the greater computing power. As an example, the human genome project initiated by Craig Venter ended with the generation of 14.8 billion base-pairs (~5× coverage) for de novo assembly ( Venter et al., 2001). Second, improved de novo assembly results are now possible by increasing the length of NGS raw reads. The first advance triggered the development of NGS technologies, including pyrosequencing ( Fakhrai-Rad et al., 2002), sequencing by synthesis ( Fuller et al., 2009), and sequencing by ligation ( Smith et al., 2010). These technologies produced a great many reads in comparison to the Sanger sequencing method, though the reads are shorter than those generated by the Sanger sequencing method, apart from those that use pyrosequencing technology. After several plant genome sequences, including A. thaliana ( The Arabidopsis Genome Initiative, 2000), Populus trichocarpa Torr. & A. Gray ex. Hook. ( Tuskan et al., 2006), Vitis vinifera L. ( Jaillon et al., 2007), and Z. mays ( Schnable et al., 2009), were deciphered by the Sanger sequencing method, the cucumber ( Cucumis sativus L.) genome was successfully assembled mainly based on sequencing by a synthesis method (commonly known as the Illumina method) ( Huang et al., 2009). Although the NGS read length is relatively short, it has been shown that plant genome sequences can be assembled based on early short-read sequences (36-bp reads) generated by NGS technology. On the other hand, the 22-Gbp plant genome of Pinus taeda L. was successfully assembled ( Zimin et al., 2014) based on pyrosequencing, implying that the de novo assembly of very large plant genomes is also possible with read lengths similar to those by the Sanger sequencing method. As more plant genome sequences continue to be sequenced based on NGS technologies at a much lower cost in comparison to the Sanger sequencing method, the short length has become a critical hurdle hindering the realization of high-quality genome assemblies. New sequencing technologies after NGS technologies, referred to as third-generation sequencing (TGS) technologies, have been developed, with high-throughput sequencing reads longer than 10 kb, much longer than those by pyrosequencing and even the Sanger sequencing method ( Terminology 1). Currently, the average length of the most recent single-molecular real-time (SMRT) sequencing method by Pacific Biosciences (PacBio) is around 10-25 kb with high accuracy ( Hon et al., 2020) and the nanopore sequencing method by Oxford Nanopore Technologies (ONT) (referred to as Nanopore hereafter) produces ultra-long reads (>100 kb) ( Amarasinghe et al., 2020).
|
Terminology 1
|
|
Next-generation sequencing (NGS) technologies: NGS technologies refer to major sequencing technologies which have been developed to overcoming the limitations of Sanger sequencing technologies addressed in the late 1990s. The first commercialized NGS technology is a pyrosequencing method known as the 454 technology method initially. In the early phase, it generated less than one million 150-bp reads but eventually could generate several million reads of which the length exceeds 1 kb (GS-Flx). This technology was officially ended in 2017. The sequencing-by-synthesis (SBS) technology was commercialized in 2007 with several million 36-bp reads per lane. This approach provided low-cost data, but each read was too short in comparison to those by the pyrosequencing method. Human genomes were successfully re-sequenced with data from this method in 2008 and 2009 (Bentley et al., 2008; Wang et al., 2008; Kim et al., 2009) and the cucumber genome was successfully assembled de novo with short-read data together with reads by the Sanger sequencing method (Huang et al., 2009). Currently this method provides a hundred million 151-bp reads per lane with the latest version of SBS technology (NovaSeq-6000). Other technologies such as sequencing-by-ligation developed by Applied Bioscience have also been commercialized, known as SOLiD (Miles et al., 2013), but disappeared after several years due to failures to address quality control issues. |
|
Third-generation sequencing (TGS) technologies: TGS technologies were developed to meet long-read requirements (more than that by the Sanger sequencing method) in a high-throughput manner (similar to NGS technologies). From the long period of stabilization of TGS technologies, the single-molecular real-time (SMRT) sequencing method by Pacific Biosciences (PacBio) and the method developed by Oxford Nanopore Technology (ONT) have been successfully commercialized, providing a large number of long reads (>10 kb). One weak point of TGS is its low base-pair accuracy; HiFi reads provided by PacBio increased this accuracy so that additional polishing is not required now (Hon et al., 2020); while Nanopore (ONT) still requires a polishing process based on NGS sequences (Amarasinghe et al., 2020) but provides much longer reads than that by PacBio. |
Based on 65 genome publications covering 72 plant genomes ( Table 1), the corresponding sequencing methods were investigated ( Fig. 1). Most of the investigated genome projects utilized multiple methods, especially for those that adopted TGS methods requiring a base pair polishing process supported by NGS technologies. Only two genomes (2.77%), A. thaliana ( The Arabidopsis Genome Initiative, 2000) and Arabidopsis lyrata (L.) O'Kane & Al-Shehbaz ( Hu et al., 2011), were sequenced with only the Sanger sequencing method ( Fig. 1, Table 1). Thirteen genomes were sequenced together with pyrosequencing and MiSeq technologies, which provide longer sequences than the typical NGS technologies; however, these methods were selected not due to the large genome (i.e., P. taeda ( Zimin et al., 2014)) but because of available optimized sequencing technologies when these projects were conducted. Since the commercialization of TGS technologies, most genomes have been sequenced with PacBio or Nanopore ( Fig. 1). Because Nanopore was commercialized later than PacBio, there have been fewer genomes deciphered using this method compared to those by PacBio ( Fig. 1). After assembling whole-genome sequences from raw reads successfully, additional processes for chromosomal-level assembly, including Hi-C ( Lieberman-Aiden et al., 2009), 10X Genomics Chromium (termed 10× hereafter) ( Weisenfeld et al., 2017), and Bionano ( Bocklandt et al., 2019), referred to as the scaffolding method hereafter, were conducted for 20 genomes (23.53%; the Rosa rugosa Thunb. genome project used both Hi-C and 10× technologies) ( Fig. 1) ( Chen et al., 2021). The most frequently used technology is Hi-C (65.00%) ( Fig. 1), which has been utilized in a wide range of genomic studies ( Kong and Zhang, 2019). The remaining scaffolding methods, including old-fashioned techniques, i.e., the BAC-end ( Osoegawa et al., 2001) and Fosmid-end ( Williams et al., 2012) sequencing methods, showed to be rarely used ( Fig. 1). Based on the ratio between N50 and the total length, recent scaffolding methods including Hi-C, 10×, and Bionano presented better efficiency of assembly with some exceptional cases, i.e., Erysimum cheiranthoides L. and Prunus davidiana Carrière ( Fig. 2). In addition, the ratio does not present a correlation with the total genome length (R 2 = 0.0397), strongly suggesting that the N50 lengths of plant genomes depend on the species ( Terminology 2).
|
Terminology 2
|
|
N50 length: The genome assembly displayed various lengths of contigs or scaffolds due to the limitation of de novo assembly, particularly the assembly of repetitive sequences. Hence, the quality of genome assembly can be estimated for the N50 length, defined as the length of the contig or scaffold for which longer contigs or scaffolds explain half of the assembled sequence. This reflects how well long sequences were assembled in comparison to average or even median lengths by focusing on relatively long assembled sequences. This approach is more effective for the assembled sequences based on long reads generated by TGS. |
CURRENT STATUS OF WHOLE GENOMES OF KOREAN ANGIOSPERM SPECIES
There are official lists of native and naturalized Korean plant species, including (1) the Database of National Species List of Korea ( Park et al., 2020a), (2) the Standard List of Korean plant species ( http://www.nature.go.kr/kpni), and (3) The Northeastern Asia Biodiversity Institute’s List of Korean Vascular Plants. All three lists suggest approximately 4,500 native and naturalized plant species in Korea, also presenting utility as a filter to select published plant genome sequences to investigate the current status of plant genomes of native and naturalized Korean plant species. We used the Northeastern Asia Biodiversity Institute’s List of Korean Vascular Plants, selecting 85 plant genomes originating from 69 taxa as Korean angiosperm species genomes from the Plant Genome Database ( https://www.plantgenome.info/), which contains available plant whole genome-sequences ( Park et al., 2021a) ( Table 1). The difference between the numbers of plant genome sequences and the taxa indicates that multiple genomes of some plant taxa were sequenced, making this a good resource to understand intraspecific variations at the level of the genome. We excluded more than 1,700 A. thaliana genomes ( Ossowski et al., 2008; Ashelford et al., 2011; Cao et al., 2011; Gan et al., 2011; Long et al., 2013; Schmitz et al., 2013; The 1001 Genomes Consortium, 2016; Zou et al., 2017) due to the extremely large number of genomes in this case. Despite the time gap between the releases of genome sequences and the corresponding publication dates, only 15.29% (13 out of 85 genomes) of genomes do not have any publication. Moreover, five species, Spirodela polyrhiza (L.) Schleid., Diospyros lotus L., Glycine soja Sieb. and Zucc., Castanea mollissima Blume, and Boehmeria nivea (L.) Gaudich., contained three genomes while six species, Panax ginseng C. A. Mey., Corylus heterophylla Fisch. ex Trautv., Setaria viridis (L.) P. Beauv., Typha latifolia L., Pyrus pyrifolia (Burm. f.) Nakai., and Ziziphus jujuba Mill., covered two genomes ( Table 1), making them good candidates for understanding intraspecific variations at the genome-wide level. Remarkably, only two genome publications with accessible genome sequences, Gastrodia elata Blume and Codonopsis lanceolata (Siebold. & Zucc.) Trautv., described the whole genome of Korean angiosperm species ( Table 1), indicating that most genomes filtered by the Korean angiosperm species list described in this paper were sequenced based on samples outside of Korea. As the importance of biological resources increases, as exemplified in the Nagoya Protocol ( Buck and Hamilton, 2011), additional efforts to obtain whole-genome sequences of these species using the samples isolated in Korea and to prepare assembled sequences (i.e., unfortunately, while the G. soja genome was sequenced in Korea ( Kim et al., 2010), no assembled sequence is available.) are required to maximize the benefits of the low sequencing costs and feasible bioinformatic analyses. Sixty-nine taxa were classified into 17 orders and 30 families. Rosales covering four families (Moraceae, Rosaceae, Rhamnaceae, and Utricaceae) contained the largest number of genomes, and Poales covered the second largest from Poaceae to Typhaceae, while Boraginales, Caryophyllales, Cucurbitales, Solanales, and Vitales had only one genome ( Fig. 3). At the family level, Rosaceae (10 taxa), Brassicaceae (7 taxa), Fabaceae (7 taxa), and Poaceae (7 taxa) contained a large number of genomes among the 30 families ( Fig. 3). This taxonomical bias was expected due to various technical factors associated with genome sequencing projects, including the genome size and ploidy. Most taxa are economically important species. Based on current sequencing technologies, especially TGS technologies, many of these problems have already been overcome; e.g., the hexaploid large genome of wheat ( Triticum aestivum L.) was sequenced and successfully assembled at the pseudo-chromosomal level ( International Wheat Genome Sequencing Consortium, 2018). Rosaceae contained ten genomes originating from five genera, specifically Prunus, Rosa, Fragaria, Malus, and Pyrus and Poaceae, had nine genomes from the six genera, displaying large taxonomic coverage ( Fig. 3, Table 1). These genomes are good candidates for understanding the genomic features of the aforementioned families.
GENOMIC STATISTICS OF 85 NATIVE AND NATURALIZED KOREAN PLANT GENOMES
The genome length and GC ratio of 85 native and naturalized Korean plant genomes were investigated, indicating average genome lengths of Araliaceae, Caryophyllaceae, and Orchidaceae of 3.20, 2.60, and 2.03 Gbp, respectively; the standard deviation of the Orchidaceae genome length was largest at 1.70 Gbp, while that of the Salicaceae genome is smallest at 16.79 Mbp ( Fig. 4). Three genomes from Orchidaceae showed that two of the three genomes were around 1 Gbp; while one was 3 Gbp in length, with Salicaceae covering two genomes from the same genus, Populus. The trend of genome size variations along with the families is congruent with the findings of earlier work ( Šmarda et al., 2014). In addition, the standard deviation of the GC ratio in Zosteraceae is extremely large, 4.05%, despite the fact that the two genomes are from the same genus, Zostera ( Table 1). Araceae, Fabaceae, and Poaceae displayed that the standard deviation of the GC ratio ranges from 1.18 to 1.25% ( Fig. 4), which is a large value in comparison to the other families. These variations in GC ratio are similar to those in a previous study that investigated variations of the GC ratio of the whole genome in a monocot species ( Šmarda et al., 2014). The number of genomes in each family is small to present a corresponding trend, therefore they provide a glimpse of family-specific genomic features through these numbers as an indicator. This indicates that further genomic studies of Korean plants are necessary.
UTILIZATION OF KOREAN ANGIOSPERM GENOME SEQUENCES
More than 1,000 plant whole genomes have been sequenced, meaning that we can investigate their characteristics in detail. However, grants and proper human resources are still required for us to present the potential or direct economic value of this resource. Genomics-assisted breeding, a good example of the utilization of whole genomes, can be conducted based on genome-wide association studies to target useful phenotypes for breeding ( Ahmar et al., 2020, 2021). To shorten the breeding time and increase the efficiency of the process, genomic editing is a viable upcoming strategy, with modifications to genetic elements to achieve helpful characteristics for humans using CRISPR-associated protein 9 ( Lee et al., 2019). Legal regulations pertaining to genetically modified organisms, considered a main suppression factor, have become positive, accepting genome-edited plants in comparison to genetically modified organisms ( Sprink et al., 2022). This development will promote the potential usage of plant whole-genome sequences because the techniques mentioned above commonly require whole genomes. Native plant resources have been utilized to develop a range of useful products, including medicines over many years due to effective compounds such as aspirin, an acetyl salicylic acid from willow bark ( Norn et al., 2009), and paclitaxel extracted from the bark of Taxus brevifolia Nutt. ( Bose et al., 2020). In addition, various natural product medicines have also been developed based on plant extracts ( Ngo et al., 2013) as well as traditional medicines, which have been utilized for several thousand years ( Ansari and Inamdar, 2010), reflecting the commercial usage of plant resources. Whole-genome sequences can be analyzed to predict useful phytocompounds because they contain all enzymes involved in biochemical synthesis theoretically ( Kang et al., 2020; Park et al., 2020b), suggesting another useful feature of Korean native plant genomes.
FURTHER DIRECTIONS FOR KOREAN ANGIOSPERM GENOME SEQUENCES
Currently, more than a hundred plant whole genomes have been sequenced per year, a rate much faster than that of ten years ago, when NGS technologies were merely utilized for the de novo assembly of plant genomes. Hence, more Korean angiosperm genomes will also be available from our own genome projects and genome projects conducted outside of Korea. We can consider two main strategies for a database for archive and utilization of Korean angiosperm genomes. First, to expand the coverage of Korean angiosperm genomes, additional Korean angiosperm genomes, especially endemic species which can be utilized commercially or which may be valuable in research, can be sequenced. Second, additional individuals or populations of Korean angiosperm species can be sequenced to understand genome-wide intraspecific variations ( Slavov et al., 2012; The 3,000 Rice Genomes Project, 2014; Gulyaev et al., 2022) as well as functional gene families ( Kim et al., 2021a, 2021b), as useful phenotypes of plant resources show differences in an intraspecific manner ( Moore et al., 2014; Aspinwall et al., 2015; Ren et al., 2020). In addition, all of these genomes can be managed under the environment of a standardized integrated platform to analyze them further, such as the web-based genomic analysis platform Galaxy ( Giardine et al., 2005; Blankenberg et al., 2010) or the Genome Information System (GeIS; https://geis.infoboss.co.kr/), which have been utilized in various genomics studies ( Lee et al., 2020; Park et al., 2020c, 2021b).
ACKNOWLEDGMENTS
This study was carried out with the support of the InfoBoss Research Grant (IBG-0042).
Fig. 1.
Sequencing methods used in whole-genome projects focusing on native and naturalized Korean plant species: the X-axis presents the sequencing methods used in genome projects with four-color tagged legends denoting their classification. The Y-axis displays the number of genomes for each method. 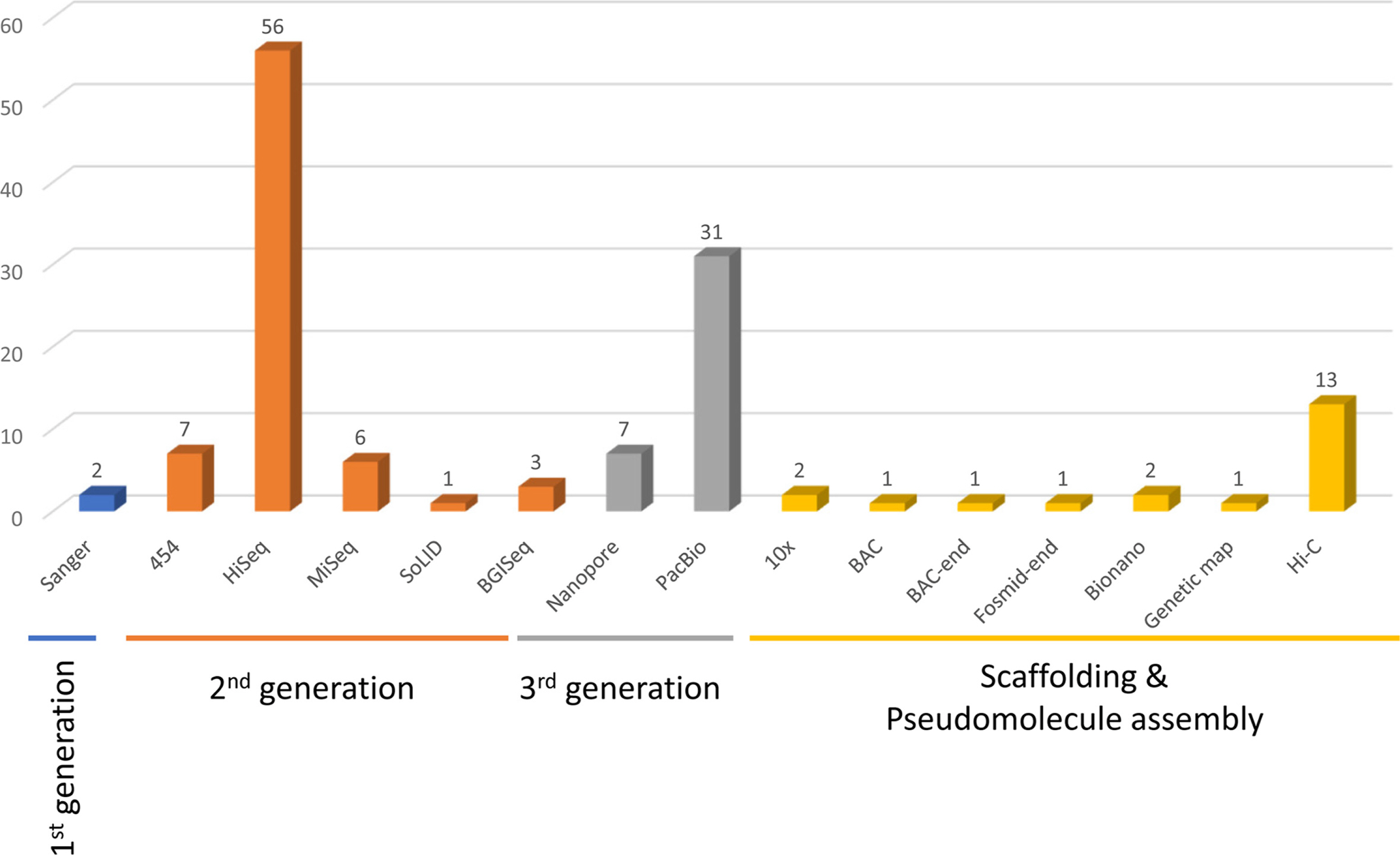
Fig. 2.
Genome assembly properties, the ratio of N50 and total lengths and total lengths of genomes using scaffolding technologies the Xaxis displays species names in a genome project with two-color tags denoting the classification of the scaffolding technologies. Bars indicate the genome length of each species and the line graph shows the ratio of N50 and the total lengths. 
Fig. 3.
Taxonomical distribution of 69 native and naturalized Korean plant taxa containing whole-genome sequences: (A) graph displaying the order-level distribution, and (B) graph showing the family-level distribution. Dotted lines represent the relationship between families and orders. 
Fig. 4.
Family-level genomic properties, total length and GC ratio: the X-axis shows families containing at least one whole genome used in this paper. Bars show the average genome length with the error bars of the standard deviation. The line represents the average GC ratio of genomes with the error bars of the standard deviation. 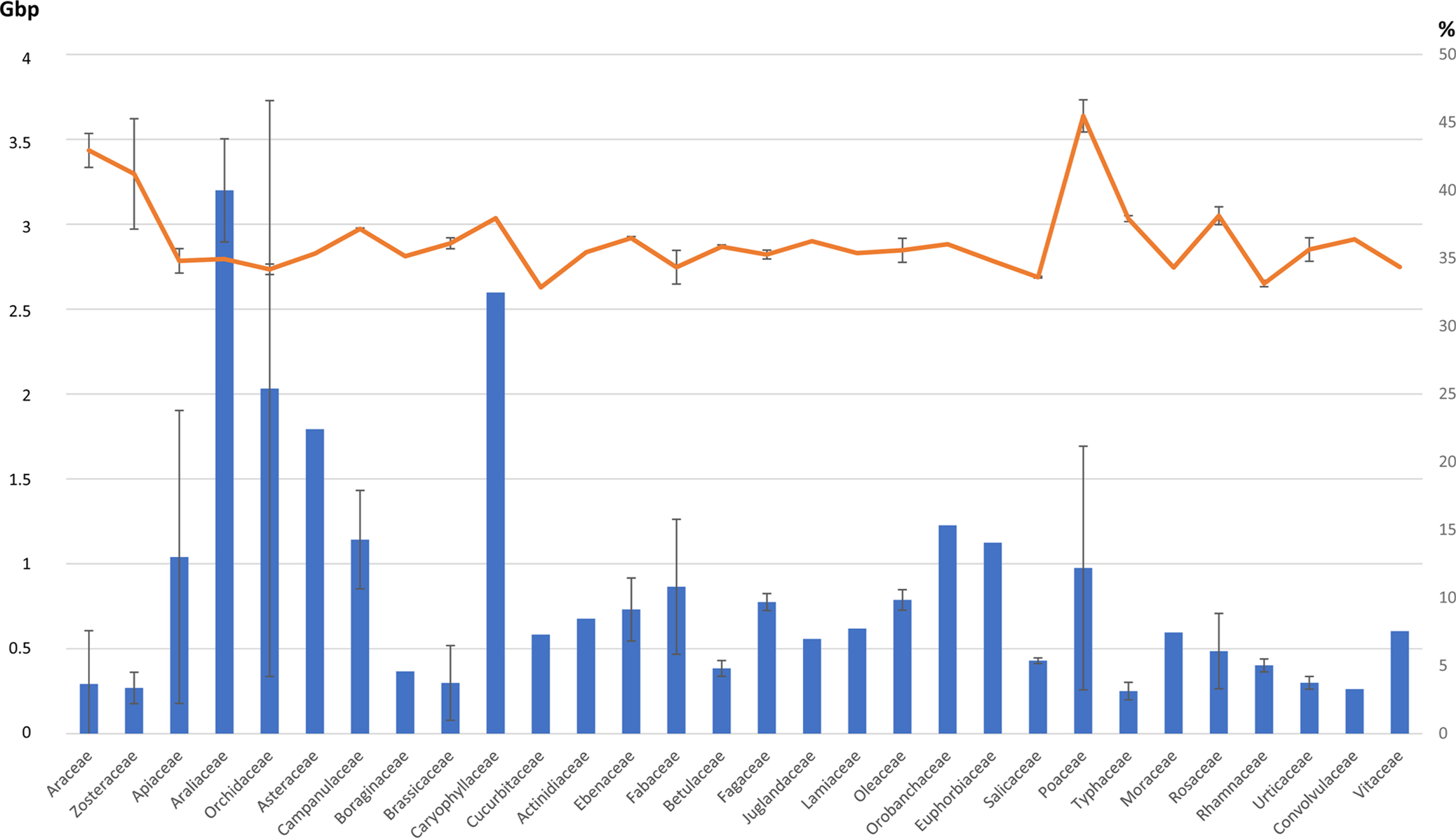
Table 1.
List of plant whole genome sequences of Korean angiosperms.
|
No. |
Order |
Family |
Species |
Cultivar/Strain |
Method |
Genome size (bp) |
No. of scaffolds |
N50 (bp) |
GC ratio (%) |
No. of genes |
Reference |
|
1 |
Alismatales |
Araceae |
Lemna minor
|
- |
HiSeq MiSeq |
763,364,415 |
6,868 |
335,126 |
44.81 |
N/A |
Van Hoeck et al. (2015)
|
|
2 |
Alismatales |
Araceae |
Spirodela polyrhiza
|
7498 |
454 BAC-end |
132,009,443 |
207 |
5,765,642 |
42.45 |
N/A |
Wang et al. (2014)
|
|
3 |
Alismatales |
Araceae |
Spirodela polyrhiza
|
9509 |
HiSeq Irys |
136,591,703 |
20 |
7,641,483 |
42.30 |
N/A |
Michael et al. (2017)
|
|
4 |
Alismatales |
Araceae |
Spirodela polyrhiza
|
Sp9504 |
N/A |
137,175,874 |
16,051 |
14,533 |
42.21 |
N/A |
Unpublished |
|
5 |
Alismatales |
Zosteraceae |
Zostera marina
|
- |
HiSeq |
203,914,448 |
2,228 |
485,578 |
38.34 |
20,450 |
Olsen et al. (2016)
|
|
6 |
Alismatales |
Zosteraceae |
Zoysia japonica
|
Nagirizaki |
HiSeq |
334,384,427 |
11,786 |
2,370,062 |
44.08 |
N/A |
Tanaka et al. (2016)
|
|
7 |
Apiales |
Apiaceae |
Centella asiatica
|
BB-174 |
10x Hi-C |
430,216,894 |
8,739 |
50,798,654 |
34.17 |
N/A |
Pootakham et al. (2021)
|
|
8 |
Apiales |
Apiaceae |
Oenanthe javanica
|
- |
HiSeq |
1,650,714,995 |
150,335 |
32,514 |
35.44 |
N/A |
Liu et al. (2021a)
|
|
9 |
Apiales |
Araliaceae |
Panax ginseng
|
- |
HiSeq |
3,414,349,854 |
83,074 |
108,708 |
34.94 |
N/A |
Xu et al. (2017)
|
|
10 |
Apiales |
Araliaceae |
Panax ginseng
|
- |
HiSeq |
2,984,993,682 |
9,845 |
3,641,815 |
34.84 |
59,352 |
Kim et al. (2018)
|
|
11 |
Asparagales |
Orchidaceae |
Gastrodia elata
|
EJP_2020-GE1012 |
HiSeq PacBio Hi-C |
1,046,143,939 |
514 |
50,595,616 |
34.27 |
N/A |
Bae et al. (2022)
|
|
12 |
Asparagales |
Orchidaceae |
Gastrodia elata f. glauca
|
- |
HiSeq |
1,060,984,162 |
3,768 |
4,911,943 |
34.52 |
N/A |
Yuan et al. (2018)
|
|
13 |
Asparagales |
Orchidaceae |
Cymbidium goeringii
|
- |
HiSeq PacBio Hi-C |
3,990,519,457 |
19,377 |
178,198,413 |
33.77 |
29,556 |
Chung et al. (2021)
|
|
14 |
Asterales |
Asteraceae |
Artemisia annua
|
- |
HiSeq 454 PacBio |
1,792,856,094 |
39,400 |
104,891 |
35.34 |
N/A |
Shen et al. (2018)
|
|
15 |
Asterales |
Campanulaceae |
Codonopsis lanceolata
|
NIHHS 239928 |
Nanopore HiSeq |
1,347,489,827 |
22,630 |
82,893 |
37.21 |
N/A |
Jang et al. (2023)
|
|
16 |
Asterales |
Campanulaceae |
Codonopsis pilosula
|
- |
N/A |
937,709,907 |
1,154 |
2,556,440 |
37.13 |
N/A |
Unpublished |
|
17 |
Boraginales |
Boraginaceae |
Lithospermum erythrorhizon
|
- |
Nanopore HiSeq |
366,684,367 |
2,451 |
315,765 |
35.15 |
32,360 |
Auber et al. (2020)
|
|
18 |
Brassicales |
Brassicaceae |
Arabidopsis lyrata
|
MN47 |
Sanger |
206,667,935 |
695 |
24,464,547 |
36.08 |
32,550 |
Hu et al. (2011)
|
|
19 |
Brassicales |
Brassicaceae |
Arabidopsis thaliana
|
Col-0 |
Sanger |
119,145,879 |
5 |
23,459,830 |
36.03 |
48,113 |
Kaul et al. (2000) |
|
20 |
Brassicales |
Brassicaceae |
Arabis glabra
|
- |
N/A |
171,129,789 |
250 |
5,712,653 |
36.01 |
N/A |
Unpublished |
|
21 |
Brassicales |
Brassicaceae |
Capsella bursa-pastoris
|
- |
HiSeq MiSeq |
268,430,517 |
8,186 |
627,605 |
35.74 |
52,528 |
Kasianov et al. (2017)
|
|
22 |
Brassicales |
Brassicaceae |
Erysimum cheiranthoides
|
- |
HiSeq PacBio Hi-C |
177,180,559 |
223 |
22,409,365 |
36.29 |
N/A |
Züst et al. (2020)
|
|
23 |
Brassicales |
Brassicaceae |
Isatis tinctoria
|
- |
N/A |
756,237,117 |
37,877 |
85,498 |
36.89 |
116,152 |
Unpublished |
|
24 |
Brassicales |
Brassicaceae |
Rorippa islandica
|
- |
N/A |
390,742,222 |
430 |
3,103,162 |
35.67 |
65,406 |
Unpublished |
|
25 |
Caryophyllales |
Caryophyllaceae |
Silene noctiflora
|
OPL-1.1 |
N/A |
2,598,026,552 |
79,767 |
59,004 |
37.95 |
N/A |
Unpublished |
|
26 |
Cucurbitales |
Cucurbitaceae |
Gynostemma pentaphyllum
|
JGL-2020 |
HiSeq BGISeq PacBio Hi-C |
582,948,444 |
578 |
50,780,587 |
32.86 |
N/A |
Huang et al. (2021)
|
|
27 |
Ericales |
Actinidiaceae |
Actinidia rufa
|
Fuchu |
HiSeq PacBio |
677,239,177 |
501 |
15,941,708 |
35.44 |
52,342 |
Tahir et al. (2022)
|
|
28 |
Ericales |
Ebenaceae |
Diospyros lotus
|
Kunsenshi |
N/A |
945,758,782 |
8,975 |
653,513 |
36.35 |
N/A |
Unpublished |
|
29 |
Ericales |
Ebenaceae |
Diospyros lotus
|
W01 |
N/A |
617,726,390 |
142 |
40,720,603 |
36.52 |
N/A |
Unpublished |
|
30 |
Ericales |
Ebenaceae |
Diospyros lotus
|
Yz01 |
N/A |
630,098,508 |
42 |
42,671,757 |
36.57 |
N/A |
Unpublished |
|
31 |
Fabales |
Fabaceae |
Amphicarpaea edgeworthii
|
AE-2020 |
BGISeq Nanopore |
299,059,313 |
24 |
27,224,559 |
32.06 |
N/A |
Liu et al. (2021b)
|
|
32 |
Fabales |
Fabaceae |
Glycine soja
|
F |
N/A |
975,918,537 |
320 |
48,777,390 |
34.78 |
57,501 |
Unpublished |
|
33 |
Fabales |
Fabaceae |
Glycine soja
|
USDA:GRIN:PI 483463 |
HiSeq PacBio Bionano |
985,259,865 |
306 |
48,820,272 |
34.94 |
N/A |
Valliyodan et al. (2019)
|
|
34 |
Fabales |
Fabaceae |
Glycine soja
|
W05 |
HiSeq PacBio Hi-C |
863,568,428 |
33,170 |
404,776 |
33.56 |
50,337 |
Xie et al. (2019)
|
|
35 |
Fabales |
Fabaceae |
Medicago ruthenica
|
Xinghe |
HiSeq PacBio Hi-C |
904,130,090 |
650 |
99,386,155 |
35.89 |
N/A |
Yin et al. (2021)
|
|
36 |
Fabales |
Fabaceae |
Vicia sativa
|
KSR5 |
HiSeq PacBio |
1,541,180,487 |
54,083 |
90,105 |
34.96 |
N/A |
Shirasawa et al. (2021b)
|
|
37 |
Fabales |
Fabaceae |
Vigna minima
|
- |
HiSeq PacBio |
486,496,346 |
2,940 |
25,346,430 |
34.19 |
N/A |
Naito et al. (2022)
|
|
38 |
Fagales |
Betulaceae |
Betula pendula
|
- |
HiSeq SOLiD |
435,914,794 |
5,644 |
239,696 |
35.74 |
86,599 |
Salojärvi et al. (2017)
|
|
39 |
Fagales |
Betulaceae |
Corylus heterophylla
|
CHET03 |
|
346,499,040 |
381 |
2,025,119 |
35.99 |
N/A |
Unpublished |
|
40 |
Fagales |
Betulaceae |
Corylus heterophylla
|
R026 |
Nanopore Hi-C |
370,750,808 |
951 |
31,328,411 |
35.84 |
N/A |
Zhao et al. (2021)
|
|
41 |
Fagales |
Fagaceae |
Castanea crenata
|
Ginyose |
HiSeq PacBio |
721,168,657 |
781 |
1,595,543 |
35.14 |
N/A |
Shirasawa et al. (2021c)
|
|
42 |
Fagales |
Fagaceae |
Castanea mollissima
|
N11-1 |
HiSeq PacBio Hi-C |
833,240,550 |
112 |
57,343,431 |
35.11 |
N/A |
Wang et al. (2020)
|
|
43 |
Fagales |
Fagaceae |
Castanea mollissima
|
Vanuxem |
454 MiSeq |
725,180,808 |
14,110 |
101,575 |
35.06 |
34,314 |
Staton et al. (2020)
|
|
44 |
Fagales |
Fagaceae |
Castanea mollissima
|
Hubei |
HiSeq PacBio |
785,529,252 |
2,707 |
944,461 |
35.21 |
36,479 |
Xing et al. (2019)
|
|
45 |
Fagales |
Fagaceae |
Quercus mongolica
|
Qm_2020SYAU |
HiSeq PacBio Hi-C |
809,993,317 |
321 |
66,735,633 |
35.84 |
N/A |
Ai et al. (2022)
|
|
46 |
Fagales |
Juglandaceae |
Juglans mandshurica
|
- |
HiSeq |
558,070,702 |
13,809 |
496,923 |
36.26 |
N/A |
Bai et al. (2018)
|
|
47 |
Lamiales |
Lamiaceae |
Perilla citriodora
|
- |
N/A |
618,797,171 |
29,924 |
2,413,193 |
35.38 |
155,867 |
Unpublished |
|
48 |
Lamiales |
Oleaceae |
Fraxinus mandshurica
|
- |
HiSeq MiSeq 454 |
830,473,688 |
297,505 |
30,144 |
34.96 |
N/A |
Sollars et al. (2017)
|
|
49 |
Lamiales |
Oleaceae |
Fraxinus sieboldiana
|
- |
HiSeq MiSeq 454 |
744,777,403 |
709,960 |
1,987 |
36.21 |
N/A |
Sollars et al. (2017)
|
|
50 |
Lamiales |
Orobanchaceae |
Phtheirospermum japonicum
|
Okayama |
N/A |
1,226,606,249 |
10,559 |
1,109,341 |
36.03 |
30,299 |
Unpublished |
|
51 |
Malpighiales |
Euphorbiaceae |
Euphorbia esula
|
- |
N/A |
1,124,886,465 |
1,633,094 |
1,035 |
34.80 |
N/A |
Unpublished |
|
52 |
Malpighiales |
Salicaceae |
Populus davidiana
|
NFU_1 |
N/A |
417,659,603 |
1,565 |
562,472 |
33.56 |
N/A |
Unpublished |
|
53 |
Malpighiales |
Salicaceae |
Populus simonii
|
- |
HiSeq PacBio |
441,407,051 |
369 |
19,598,675 |
33.65 |
N/A |
Wu et al. (2020)
|
|
54 |
Poales |
Poaceae |
Brachypodium sylvaticum
|
- |
N/A |
358,283,154 |
629 |
38,764,466 |
46.38 |
50,263 |
Unpublished |
|
55 |
Poales |
Poaceae |
Echinochloa crus-galli
|
STB08 |
HiSeq PacBio |
1,486,609,408 |
4,534 |
1,802,240 |
45.70 |
N/A |
Guo et al. (2017)
|
|
56 |
Poales |
Poaceae |
Eleusine indica
|
HZ-2018 |
HiSeq |
492,270,386 |
24,072 |
233,459 |
44.42 |
N/A |
Zhang et al. (2019)
|
|
57 |
Poales |
Poaceae |
Miscanthus sacchariflorus
|
- |
HiSeq |
2,074,815,175 |
105,321 |
37,711 |
45.94 |
N/A |
De Vega et al. (2021)
|
|
58 |
Poales |
Poaceae |
Miscanthus sinensis
|
- |
HiSeq Hi-C |
2,079,430,866 |
14,431 |
88,510,541 |
45.77 |
89,486 |
Mitros et al. (2020)
|
|
59 |
Poales |
Poaceae |
Setaria viridis
|
A10 |
HiSeq pacBio |
395,731,502 |
14 |
46,702,114 |
46.17 |
52,459 |
Mamidi et al. (2020)
|
|
60 |
Poales |
Poaceae |
Setaria viridis
|
ME034V |
HiSeq Nanopore |
397,031,521 |
9 |
46,382,547 |
45.99 |
N/A |
Thielen et al. (2020)
|
|
61 |
Poales |
Poaceae |
Themeda triandra
|
sf006 |
N/A |
889,201,528 |
3,179 |
600,859 |
46.24 |
N/A |
Unpublished |
|
62 |
Poales |
Poaceae |
Zizania latifolia
|
HSD2 |
HiSeq |
603,989,347 |
4,522 |
604,864 |
42.72 |
N/A |
Guo et al. (2015)
|
|
63 |
Poales |
Typhaceae |
Typha latifolia
|
L0001 |
N/A |
214,326,387 |
81 |
14,472,745 |
37.76 |
N/A |
Unpublished |
|
64 |
Poales |
Typhaceae |
Typha latifolia
|
SDW-2020 |
HiSeq PacBio |
287,194,721 |
1,158 |
8,705,712 |
38.07 |
N/A |
Widanagama et al. (2022)
|
|
65 |
Rosales |
Moraceae |
Ficus erecta
|
- |
HiSeq PacBio Genetic map |
595,834,738 |
2,455 |
697,200 |
34.32 |
111,921 |
Shirasawa et al. (2020)
|
|
66 |
Rosales |
Rosaceae |
Fragaria nipponica
|
- |
454 HiSeq |
206,414,979 |
215,024 |
1,275 |
38.44 |
87,803 |
Hirakawa et al. (2014)
|
|
67 |
Rosales |
Rosaceae |
Fragaria orientalis
|
- |
454 HiSeq |
214,184,046 |
323,163 |
722 |
38.11 |
99,674 |
Hirakawa et al. (2014)
|
|
68 |
Rosales |
Rosaceae |
Malus baccata
|
- |
HiSeq |
703,023,564 |
47,474 |
743,357 |
38.13 |
45,900 |
Chen et al. (2019)
|
|
69 |
Rosales |
Rosaceae |
Prunus yedoensis var. nudiflora*
|
- |
HiSeq PacBio Fosmid-end |
319,209,792 |
4,016 |
145,140 |
37.65 |
41,294 |
Baek et al. (2018)
|
|
70 |
Rosales |
Rosaceae |
Prunus yeonesis
|
- |
HiSeq PacBio |
690,105,700 |
4,571 |
918,183 |
37.93 |
N/A |
Shirasawa et al. (2019)
|
|
71 |
Rosales |
Rosaceae |
Prunus davidiana
|
ST |
HiSeq PacBio Hi-C |
243,913,910 |
127 |
28,110,464 |
37.55 |
N/A |
Tan et al. (2021)
|
|
72 |
Rosales |
Rosaceae |
Pyrus pyrifolia
|
Nijisseiki |
BGISeq PacBio |
503,888,133 |
114 |
7,676,629 |
37.34 |
N/A |
Shirasawa et al. (2021a)
|
|
73 |
Rosales |
Rosaceae |
Pyrus pyrifolia
|
Cuiguan |
HiSeq PacBio |
541,340,367 |
428 |
27,968,327 |
37.36 |
42,559 |
Gao et al. (2021)
|
|
74 |
Rosales |
Rosaceae |
Rosa lucieae
|
- |
N/A |
786,105,075 |
500,476 |
10,695 |
38.73 |
N/A |
Unpublished |
|
75 |
Rosales |
Rosaceae |
Rosa multiflora
|
- |
HiSeq MiSeq |
739,637,845 |
83,189 |
90,830 |
38.91 |
67,380 |
Nakamura et al. (2018)
|
|
76 |
Rosales |
Rosaceae |
Rosa rugosa
|
- |
PacBio 10x Hi-C |
401,223,225 |
43 |
61,014,187 |
39.31 |
39,704 |
Chen et al. (2021)
|
|
77 |
Rosales |
Rhamnaceae |
Ziziphus jujuba
|
Dongzao |
HiSeq BAC |
437,753,511 |
4,789 |
25,259,912 |
33.40 |
37,526 |
Liu et al. (2014)
|
|
78 |
Rosales |
Rhamnaceae |
Ziziphus jujuba
|
Junzao |
HiSeq |
362,583,438 |
47 |
27,412,306 |
32.95 |
N/A |
Huang et al. (2016)
|
|
79 |
Rosales |
Rhamnaceae |
Ziziphus jujuba var. spinosa
|
AT0 |
HiSeq PacBio Hi-C |
405,636,987 |
538 |
30,278,369 |
33.07 |
24,146 |
Shen et al. (2021)
|
|
80 |
Rosales |
Urticaceae |
Boehmeria nivea
|
- |
|
344,616,830 |
12,775 |
1,094,501 |
35.25 |
N/A |
Unpublished |
|
81 |
Rosales |
Urticaceae |
Boehmeria nivea
|
ZZ1 |
HiSeq |
316,026,324 |
330 |
17,838,734 |
35.20 |
N/A |
Liu et al. (2018)
|
|
82 |
Rosales |
Urticaceae |
Boehmeria nivea
|
Zhongsizhu |
HiSeq Nanopore PacBio |
266,599,076 |
154,955 |
48,874 |
36.94 |
N/A |
Wang et al. (2021a)
|
|
83 |
Rosales |
Urticaceae |
Boehmeria nivea var. tenacissima
|
- |
HiSeq Nanopore PacBio |
270,213,153 |
135 |
19,552,154 |
35.19 |
N/A |
Wang et al. (2021a)
|
|
84 |
Solanales |
Convolvulaceae |
Cuscuta australis
|
|
HiSeq PacBio |
262,630,465 |
218 |
3,625,894 |
36.40 |
18,157 |
Sun et al. (2018)
|
|
85 |
Vitales |
Vitaceae |
Vitis amurensis
|
IBCAS1988 |
HiSeq PacBio Hi-C Bionano |
603,559,200 |
3,040 |
26,085,413 |
34.36 |
N/A |
Wang et al. (2021b)
|
LITERATURE CITED
Ai, W. Liu, Y. Mei, M. Zhang, X. Tan, E. Liu, H. Han, X. Zhan, H and Lu, X. 2022. A chromosome-scale genome assembly of the Mongolian oak ( Quercus mongolica). Molecular Ecology Resources 22: 2396-2410.    Alberts, B. Johnson, A. Lewis, J. Raff, M. Roberts, K and Walter, P. 2002. Molecular Biology of the Cell. 4th ed. Garland Science, New York. 1616 pp.
Ansari, J. A and Inamdar, NN. 2010. The promise of traditional medicines. International Journal of Pharmacology 6: 808-812. Ashelford, K. Eriksson, M. E. Allen, C. M. D'Amore, R. Johansson, M. Gould, P. Kay, S. Millar, A. J. Hall, N and Hall, A. 2011. Full genome re-sequencing reveals a novel circadian clock mutation in. Arabidopsis. Genome Biology 12: R28.
Aspinwall, M. J. Loik, M. E. Resco de Dios, V. Tjoelker, M. G. Payton, P. R and Tissue, D. T. 2015. Utilizing intraspecific variation in phenotypic plasticity to bolster agricultural and forest productivity under climate change. Plant, Cell & Environment 38: 1752-1764. Bae, E.-K. An, C. Kang, M.-J. Lee, S.-A. Lee, S. J. Kim, K.-T and Park, E.-J. 2022. Chromosome-level genome assembly of the fully mycoheterotrophic orchid Gastrodia elata
. G3 Genes|Genomes|Genetics 12: jkab433. Baek, S. Choi, K. Kim, G.-B. Yu, H.-J. Cho, A. Jang, H. Kim, C. Kim, H.-J. Chang, K. S. Kim, J.-H and Mun, J.-H. 2018. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biology 19: 127.  Bai, W.-N. Yan, P.-C. Zhang, B.-W. Woeste, K. E. Lin, K and Zhang, D.- Y. 2018. Demographically idiosyncratic responses to climate change and rapid Pleistocene diversification of the walnut genus Juglans (Juglandaceae) revealed by wholegenome sequences. New Phytologist 217: 1726-1736. Bentley, D. R. Balasubramanian, S. Swerdlow, H. P. Smith, G. P. Milton, J. Brown, C. G. Hall, K. P. Evers, D. J. Barnes, C. L. Bignell, H. R. Boutell, J. M. Bryant, J. Carter, R. J. Keira Cheetham, R. Cox, A. J. Ellis, D. J. Flatbush, M. R. Gormley, N. A. Humphray, S. J. Irving, L. J. Karbelashvili, M. S. Kirk, S. M. Li, H. Liu, X. Maisinger, K. S. Murray, L. J. Obradovic, B. Ost, T. Parkinson, M. L. Pratt, M. R. Rasolonjatovo, I. M. J. Reed, M. T. Rigatti, R. Rodighiero, C. Ross, M. T. Sabot, A. Sankar, S. V. Scally, A. Schroth, G. P. Smith, M. E. Smith, V. P. Spiridou, A. Torrance, P. E. Tzonev, S. S. Vermaas, E. H. Walter, K. Wu, X. Zhang, L. Alam, M. D. Anastasi, C. Aniebo, I. C. Bailey, D. M. D. Bancarz, I. R. Banerjee, S. Barbour, S. G. Baybayan, P. A. Benoit, V. A. Benson, K. F. Bevis, C. Black, P. J. Boodhun, A. Brennan, J. S. Bridgham, J. A. Brown, R. C. Brown, A. A. Buermann, D. H. Bundu, A. A. Burrows, J. C. Carter, N. P. Castillo, N. Catenazzi, M. C. E. Chang, S. Cooley, R. N. Crake, N. R. Dada, O. O. Diakoumakos, K. D. Dominguez-Fernandez, B. Earnshaw, D. J. Egbujor, U. C. Elmore, D. W. Etchin, S. S. Ewan, M. R. Fedurco, M. Fraser, L. J. Fuentes Fajardo, K. V. Scott Furey, W. George, D. Gietzen, K. J. Goddard, C. P. Golda, G. S. Granieri, P. A. Green, D. E. Gustafson, D. L. Hansen, N. F. Harnish, K. Haudenschild, C. D. Heyer, NI. Hims, M. M. Ho, J. T. Horgan, A. M. Hoschler, K. Hurwitz, S. Ivanov, D. V. Johnson, M. Q. James, T. Huw Jones, T. A. Kang, G.-D. Kerelska, T. H. Kersey, A. D. Khrebtukova, I. Kindwall, A. P. Kingsbury, Z. Kokko-Gonzales, P. I. Kumar, A. Laurent, M. A. Lawley, C. T. Lee, S. E. Lee, X. Liao, A. K. Loch, J. A. Lok, M. Luo, S. Mammen, R. M. Martin, J. W. McCauley, P. G. McNitt, P. Mehta, P. Moon, K. W. Mullens, J. W. Newington, Taksina. Ning, Zemin. Ng, Bee Ling. Novo, Sonia M. O’Neill, Michael J. Osborne, M. A. Osnowski, A. Ostadan, O. Paraschos, L. L. Pickering, L. Pike, A. C. Pike, A. C. Chris Pinkard, D. Pliskin, D. P. Podhasky, J. Quijano, V. J. Raczy, C. Rae, V. H. Rawlings, S. R. Rodriguez, A. C. Roe, P. M. Rogers, J. Bacigalupo, M. C. Rogert. Romanov, N. Romieu, A. Roth, R. K. Rourke, N. J. Ruediger, S. T. Rusman, E. Sanches-Kuiper, R. M. Schenker, M. R. Seoane, J. M. Shaw, R. J. Shiver, M. K. Short, S. W. Sizto, NL. Sluis, J. P. Smith, M. A. Sohna, J. E. S. Spence, E. J. Stevens, K. Sutton, N. Szajkowski, L. Tregidgo, C. L. Turcatti, G. vandeVondele, S. Verhovsky, Y. Virk, S. M. Wakelin, S. Walcott, G. C. Wang, J. Worsley, G. J. Yan, J. Yau, L. Zuerlein, M. Rogers, J. Mullikin, J. C. Hurles, M. E. McCooke, N. J. West, J. S. Oaks, F. L. Lundberg, P. L. Klenerman, D. Durbin, R and Smith, A. J. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53-59. Bocklandt, S. Hastie, A and Cao, H. 2019. Bionano genome mapping: high-throughput, ultra-long molecule genome analysis system for precision genome assembly and haploid-resolved structural variation discovery. Single Molecule and Single Cell Sequencing. Advances in Experimental Medicine and Biology. 1129: Suzuki, Y (ed.), Springer, Singapore. 97-118. Buck, M and Hamilton, C. 2011. The Nagoya Protocol on access to genetic resources and the fair and equitable sharing of benefits arising from their utilization to the Convention on Biological Diversity. Review of European Community & International Environmental Law 20: 47-61. Cao, J. Schneeberger, K. Ossowski, S. Günther, T. Bender, S. Fitz, J. Koenig, D. Lanz, C. Stegle, O. Lippert, C. Wang, X. Ott, F. Müller, J. Alonso-Blanco, C. Borgwardt, K. Schmid, K. J and Weigel, D. 2011. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nature Genetics 43: 956-963. Chen, F. Su, L. Hu, S. Xue, J.-Y. Liu, H. Liu, G. Jiang, Y. Du, J. Qiao, Y. Fan, Y. Liu, H. Yang, Q. Lu, W. Shao, Z.-Q. Zhang, J. Zhang, L. Chen, F and Ceng, Z.-M. (Max). 2021. A chromosome- level genome assembly of rugged rose ( Rosa rugosa) provides insights into its evolution, ecology, and floral characteristics. Horticulture Research 8: 141. Chen, X. Li, S. Zhang, D. Han, M. Jin, X. Zhao, C. Wang, S. Xing, L. Ma, J. Ji, J and An, N. 2019. Sequencing of a wild apple ( Malus baccata) genome unravels the differences between cultivated and wild apple species regarding disease resistance and cold tolerance. G3: Genes, Genomes, Genetics 9: 2051-2060. Chung, G. Y. Jang, H.-D. Chang, K. S. Choi, H. J. Kim, Y.-S. Kim, H.-J and Son, D. C. 2023. A checklist of endemic plants on the Korean Peninsula II. Korean Journal of Plant Taxonomy 53: 79-101. Chung, O. Kim, J. Bolser, D. Kim, H.-M. Jun, J. H. Choi, J.-P. Jang, H.- D. Cho, Y. S. Bhak, J and Kwak, M. 2021. A chromosome- scale genome assembly and annotation of the spring orchid ( Cymbidium goeringii). Molecular Ecology Resources 22: 1168-1177. Fakhrai-Rad, H. Pourmand, N and Ronaghi, M. 2002. Pyrosequencing: an accurate detection platform for single nucleotide polymorphisms. Human Mutation 19: 479-485. Fleischmann, R. D. Adams, M. D. White, O. Clayton, R. A. Kirkness, E. F. Kerlavage, A. R. Bult, C. J. Tomb, J. F. Dougherty, B. A. Merrick, J. M. McKenney, K. Sutton, G. Fitzhugh, C. Fields, W. Gocayne, J. D. Scott, J. Shirley, R. Liu, L.-L. Glodek, A. Kelley, J. M. Weidman, J. F. Phillips, C. A. Spriggs, T. Hedblom, E. Cotton, M. D. Utterback, T. R. Hanna, M. C. Nguyen, D. T. Saudek, D. M. Brandon, R. C. Fine, L. D. Fritchman, J. L. Fuhrmann, J. L. Geoghagen, N. S. M. Gnehm, C. L. McDonald, L. A. Small, K. V. Fraser, C. Smith, H. O and Craig Venter, J. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269: 496-512. Fuller, C. W. Middendorf, L. R. Benner, S. A. Church, G. M. Harris, T. Huang, X. Jovanovich, S. B. Nelson, J. R. Schloss, J. A. Schwartz, D. C and Vezenov, D. V. 2009. The challenges of sequencing by synthesis. Nature Biotechnology 27: 1013-1023. Gan, X. Stegle, O. Behr, J. Steffen, J. G. Drewe, P. Hildebrand, K. L. Lyngsoe, R. Schultheiss, S. J. Osborne, E. J. Sreedharan, V. T. Kahles, A. Bohnert, R. Jean, G. Derwent, P. Kersey, P. Belfield, E. J. Harberd, N. P. Kemen, E. Toomajian, C. Kover, P. X. Clark, R. M. Rätsch, G and Mott, R. 2011. Multiple reference genomes and transcriptomes for Arabidopsis thaliana
. Nature 477: 419-423. Gao, Y. Yang, Q. Yan, X. Wu, X. Yang, F. Li, J. Wei, J. Ni, J. Ahmad, M. Bai, S and Teng, Y. 2021. High-quality genome assembly of‘Cuiguan’pear ( Pyrus pyrifolia) as a reference genome for identifying regulatory genes and epigenetic modifications responsible for bud dormancy. Horticulture Research 8: 197. Giardine, B. Riemer, C. Hardison, R. C. Burhans, R. Elnitski, L. Shah, P. Zhang, Y. Blankenberg, D. Albert, I. Taylor, J. Miller, W. Kent, W. J and Nekrutenko, A. 2005. Galaxy: a platform for interactive large-scale genome analysis. Genome Research 15: 1451-1455. Goffeau, A. Barrell, B. G. Bussey, H. Davis, R. W. Dujon, B. Feldmann, H. Galibert, F. Hoheisel, J. D. Jacq, C. Johnston, M. Louis, E. J. Mewes, H. W. Murakami, Y. Philippsen, P. Tettelin, H and Oliver, S. G. 1996. Life with 6000 genes. Science 274: 546-567. Guo, L. Qiu, J. Han, Z. Ye, Z. Chen, C. Liu, C. Xin, X. Ye, C.-Y. Wang, Y.-Y. Xie, H. Wang, Y. Bao, J. Tang, S. Xu, J. Gui, Y. Fu, F. Wang, W. Zhang, X. Zhu, Q. Guang, X. Wang, C. Cui, H. Cai, D. Ge, S. Tuskan, G. A. Yang, X. Qian, Q. He, S. Y. Wang, J. Zhou, X.-P and Fan, L. 2015. A host plant genome ( Zizania latifolia) after a century-long endophyte infection. The Plant Journal 83: 600-609. Guo, L. Qiu, J. Ye, C. Jin, G. Mao, L. Zhang, H. Yang, X. Peng, Q. Wang, Y. Jia, L. Lin, Z. Li, G. Fu, F. Liu, C. Chen, L. Shen, E. Wang, W. Chu, Q. Wu, D. Wu, S. Xia, C. Zhang, Y. Zhou, X. Wang, L. Wu, L. Song, W. Wang, Y. Shu, Q. Aoki, D. Yumoto, E. Yokota, T. Miyamoto, K. Okada, K. Kim, D.-S. Cai, D. Zhang, C. Lou, Y. Qian, Q. Yamaguchi, H. Yamane, H. Kong, C.-H. Timko, M. P. Bai, L and Fan, L. 2017.
Echinochloa crus-galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nature Communications 8: 1031. Hirakawa, H. Shirasawa, K. Kosugi, S. Tashiro, K. Nakayama, S. Yamada, M. Kohara, M. Watanabe, A. Kishida, Y. Fujishiro, T. Tsuruoka, H. Minami, C. Sasamoto, S. Kato, M. Nanri, K. Komaki, A. Yanagi, T. Guoxin, Q. Maeda, F. Ishikawa, M. Kuhara, S. Sato, S. Tabata, S and Isobe, S. 2014. Dissection of the octoploid strawberry genome by deep sequencing of the genomes of Fragaria species. DNA Research 21: 169-181. Hon, T. Mars, K. Young, G. Tsai, Y.-C. Karalius, J. W. Landolin, J. M. Maurer, N. Kudrna, D. Hardigan, M. A. Steiner, C. C. Knapp, S. J. Ware, D. Shapiro, B. Peluso, P and Rank, D. R. 2020. Highly accurate long-read HiFi sequencing data for five complex genomes. Scientific Data 7: 399. Hu, T. T. Pattyn, P. Bakker, E. G. Cao, J. Cheng, J.-F. Clark, R. M. Fahlgren, N. Fawcett, J. A. Grimwood, J. Gundlach, H. Haberer, G. Hollister, J. D. Ossowski, S. Ottilar, R. P. Salamov, A. A. Schneeberger, K. Spannagl, M. Wang, X. Yang, L. Nasrallah, M. E. Bergelson, J. Carrington, J. C. Gaut, B. S. Schmutz, J. Mayer, K. F. X. Van de Peer, Y. Grigoriev, I. V. Nordborg, M. Weigel, D and Guo, Y.-L. 2011. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nature Genetics 43: 476-481. Huang, J. Zhang, C. Zhao, X. Fei, Z. Wan, K. Zhang, Z. Pang, X. Yin, X. Bai, Y. Sun, X. Gao, L. Li, R. Zhang, J and Li, X. 2016. The jujube genome provides insights into genome evolution and the domestication of sweetness/acidity taste in fruit trees. PLoS Genetics 12: e1006433. Huang, S. Li, R. Zhang, Z. Li, L. Gu, X. Fan, W. Lucas, W. J. Wang, X. Xie, B. Ni, P. Ren, Y. Zhu, H. Li, J. Lin, K. Jin, W. Fei, Z. Li, G. Staub, J. Kilian, A. van der Vossen, E. A. G. Wu, Y. Guo, J. He, J. Jia, Z. Ren, Y. Tian, G. Lu, Y. Ruan, J. Qian, W. Wang, M. Huang, Q. Li, B. Xuan, Z. J. Cao, Asan. Wu, Z. Zhang, J. Cai, Q. Bai, Y. Zhao, B. Han, Y. Li, Y. Li, X. Wang, S. Shi, Q. Liu, S. Cho, W. K. Kim, J.-Y. Xu, Y. Heller-Uszynska, K. Miao, H. Cheng, Z. Zhang, S. Wu, J. Yang, Y. Kang, H. Li, M. Liang, H. Ren, X. Shi, Z. Wen, M. Jian, M. Yang, H. Zhang, G. Yang, Z. Chen, R. Liu, S. Li, J. Ma, L. Liu, H. Zhou, Y. Zhao, J. Fang, X. Li, G. Fang, L. Li, Y. Liu, D. Zheng, H. Zhang, Y. Qin, N. Li, Z. Yang, G. Yang, S. Bolund, L. Kristiansen, K. Zheng, H. Li, S. Zhang, X. Yang, H. Wang, J. Sun, R. Zhang, B. Jiang, S. Wang, J. Du, Y and Li, S. 2009. The genome of the cucumber, Cucumis sativus L. Nature Genetics 41: 1275-1281. Hutchison, C. A III. Chuang, R.-Y. Noskov, V. N. Assad-Garcia, N. Deerinck, T. J. Ellisman, M. H. Gill, J. Kannan, K. Karas, B. J. Ma, L. Pelletier, J. F. Qi, Z.-Q. Alexander Richter, R. Strychalski, E. A. Sun, L. Suzuki, Y. Tsvetanova, B. Wise, K. S. Smith, H. O. Glass, J. I. Merryman, C. Gibson, D. G and Craig Venter, J. 2016. Design and synthesis of a minimal bacterial genome. Science 351: aad6253. International Wheat Genome Sequencing Consortium (IWGSC). 2018. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361: eaar7191. Jaillon, O. Aury, J.-M. Noel, B. Policriti, A. Clepet, C. Cassagrande, A. Choisne, N. Aubourg, S. Vitulo, N. Jubin, C. Vezzi, A. Legeai, F. Hugueney, P. Dasilva, C. Horner, D. Mica, E. Jublot, D. Poulain, J. Bruyère, C. Billault, A. Segurens, B. Gouyvenoux, M. Ugarte, E. Cattonaro, F. Anthouard, V. Vico, V. Del Fabbro, C. Alaux, M. Di Gaspero, G. Dumas, V. Felice, N. Paillard, S. Juman, I. Moroldo, M. Scalabrin, S. Canaguier, A. Le Clainche, I. Malacrida, G. Durand, E. Pesole, G. Laucou, V. Chatelet, P. Merdinoglu, D. Delledonne, M. Pezzotti, M. Lecharny, A. Scarpelli, C. Artiguenave, F. Enrico Pè, M. Valle, G. Morgante, M. Caboche, M. Adam-Blondon, A.-F. Weissenbach, J. Quétier, F. Wincker, P and French-Italian Public Consortium for Grapevine Genome Characterization. 2007. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449: 463-467. Jang, W. Kang, J.-N. Jo, I.-H. Lee, S.-M. Park, G-H and Kim, C-K. 2023. The chromosome-level genome assembly of lance asiabell ( Codonopsis lanceolata), a medicinal and vegetable plant of the Campanulaceae family. Frontiers in Genetics 14: 1100819. Jones-Rhoades, M. W. Bartel, D. P and Bartel, B. 2006. MicroR-NAs and their regulatory roles in plants. Annual Review of Plant Biology 57: 19-53. Kang, S.-H. Pandey, R. P. Lee, C.-M. Sim, J.-S. Jeong, J.-T. Choi, B.-S. Jung, M. Ginzburg, D. Zhao, K. Won, S. Y. Oh, T.-J. Yu, Y. Kim, N.-H. Lee, O. R. Lee, T.-H. Bashyal, P. Kim, T.-S. Lee, W.-H. Hawkins, C. Kim, C.-K. Kim, J. S. Ahn, B. O. Rhee, S. Y and Sohng, J. K. 2020. Genome-enabled discovery of anthraquinone biosynthesis in Senna tora
. Nature Communications 11: 5875. Kasianov, A. S. Klepikova, A. V. Kulakovskiy, I. V. Gerasimov, E. S. Fedotova, A. V. Besedina, E. G. Kondrashov, A. S. Logacheva, M. D and Penin, A. A. 2017. High-quality genome assembly of Capsella bursa-pastoris reveals asymmetry of regulatory elements at early stages of polyploid genome evolution. The Plant Journal 91: 278-291. Kim, J.-I. Ju, Y. S. Park, H. Kim, S. Lee, S. Yi, J.-H. Mudge, J. Miller, N. A. Hong, D. Bell, C. J. Kim, H.-S. Chung, I.-S. Lee, W.-C. Lee, J.-S. Seo, S.-H. Yun, J.-Y. Woo, H. N. Lee, H. Suh, D. Lee, S. Kim, H.-J. Yavartanoo, M. Kwak, M. Zheng, Y. Lee, M. K. Park, H. Kim, J. Y. Gokcumen, O. Mills, R. E. Zaranek, A. W. Thakuria, J. Wu, X. Kim, R. W. Huntley, J. J. Luo, S. Schroth, G. P. Wu, T. D. Kim, H. Yang, K.-S. Park, W.-Y. Kim, H. Church, G. M. Lee, C. Kingsmore, S. F and Seo, J.-S. 2009. A highly annotated whole-genome sequence of a Korean individual. Nature 460: 1011-1015. Kim, M. Y. Lee, S. Van, K. Kim, T.-H. Jeong, S.-C. Choi, I.-Y. Kim, D.- S. Lee, Y.-S. Park, D. Ma, J. Kim, W.-Y. Kim, B.-C. Park, S. Lee, K.-A. Kim, D. H. Kim, K. H. Shin, J. H. Jang, Y. E. Kim, K. D. Liu, W. X. Chaisan, T. Kang, Y. J. Lee, Y.-H. Kim, K.- H. Moon, J.-K. Schmutz, J. Jackson, S. A. Bhak, J and Lee, S.-H. 2010. Whole-genome sequencing and intensive analysis of the undomesticated soybean (G lycine soja Sieb. and Zucc.) genome. Proceedings of the National Academy of Sciences 107: 22032-22037. Kim, N.-H. Jayakodi, M. Lee, S.-C. Choi, B.-S. Jang, W. Lee, J. Kim, H. H. Waminal, N. E. Lakshmanan, M. van Nguyen, B. Lee, Y. S. Park, H.-S. Koo, H. J. Park, J. Y. Perumal, S. Joh, H. J. Lee, H. Kim, J. Kim, I. S. Kim, K. Koduru, L. Kang, K. B. Sung, S. H. Yu, Y. Park, D. S. Choi, D. Seo, E. Kim, S. Kim, Y.-C. Hyun, D. Y. Park, Y.-I. Kim, C. Lee, T.-H. Kim, H. U. Soh, M. S. Lee, Y. In, J. G. Kim, H.-S. Kim, Y.-M. Yang, D.-C. Wing, R. A. Lee, D.- Y. Paterson, A. H and Yang, T.-J. 2018. Genome and evolution of the shade-requiring medicinal herb Panax ginseng
. Plant Biotechnology Journal 16: 1904-1917. Lewin, H. A. Robinson, G. E. Kress, W. J. Baker, W. J. Coddington, J. Crandall, K. A. Durbin, R. Edwards, S. V. Forest, F. Gilbert, M. T. P. Goldstein, M. M. Grigoriev, I. V. Hackett, K. J. Haussler, D. Jarvis, E. D. Johnson, W. E. Patrinos, A. Richards, S. Castilla-Rubio, J. C. van Sluys, M.-A. Soltis, P. S. Xu, X. Yang, H and Zhang, G. 2018. Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences of the United States of America 115: 4325-4333. Lieberman-Aiden, E. van Berkum, NL. Williams, L. Imakaev, M. Ragoczy, T. Telling, A. Amit, I. Lajoie, B. R. Sabo, P. J. Dorschner, M. O. Sandstrom, R. Bernstein, B. Bender, M. A. Groudine, M. Gnirke, A. Stamatoyannopoulos, J. Mirny, L. A. Lander, E. S and Dekker, J. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289-293. Liu, C. Zeng, L. Zhu, S. Wu, L. Wang, Y. Tang, S. Wang, H. Zheng, X. Zhao, J. Chen, X. Dai, Q and Liu, T. 2018. Draft genome analysis provides insights into the fiber yield, crude protein biosynthesis, and vegetative growth of domesticated ramie ( Boehmeria nivea L. Gaud). DNA Research 25: 173-181. Liu, J.-X. Jiang, Q. Tao, J.-P. Feng, K. Li, T. Duan, A.-Q. Wang, H. Xu, Z.-S. Liu, H and Xiong, A-S. 2021a. Integrative genome, transcriptome, microRNA, and degradome analysis of water dropwort ( Oenanthe javanica) in response to water stress. Horticulture Research 8: 262. Liu, M.-J. Zhao, J. Cai, Q.-L. Liu, G.-C. Wang, J.-R. Zhao, Z.-H. Liu, P. Dai, L. Yan, G. Wang, W.-J. Li, X.-S. Chen, Y. Sun, Y.-D. Liu, Z.-G. Lin, M.-J. Xiao, J. Chen, Y.-Y. Li, X.-F. Wu, B. Ma, Y. Jian, J.-B. Yang, W. Yuan, Z. Sun, X.-C. Wei, Y.-L. Yu, L.- L. Zhang, C. Liao, S.-G. He, R.-J. Guang, X.-M. Wang, Z. Zhang, Y.-Y and Luo, L.-H. 2014. The complex jujube genome provides insights into fruit tree biology. Nature Communications 5: 5315. Liu, Y. Zhang, X. Han, K. Li, R. Xu, G. Han, Y. Cui, F. Fan, S. Seim, I. Fan, G. Li, G and Wan, S. 2021b. Insights into amphicarpy from the compact genome of the legume Amphicarpaea edgeworthii
. Plant Biotechnology Journal 19: 952-965. Long, Q. Rabanal, F. A. Meng, D. Huber, C. D. Farlow, A. Platzer, A. Zhang, Q. Vilhjálmsson, B. J. Korte, A. Nizhynska, V. Voronin, V. Korte, P. Sedman, L. Mandáková, T. Lysak, M. A. Seren, Ü. Hellmann, I and Nordborg, M. 2013. Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nature Genetics 45: 884-890. Mamidi, S. Healey, A. Huang, P. Grimwood, J. Jenkins, J. Barry, K. Sreedasyam, A. Shu, S. Lovell, J. T. Feldman, M. Wu, J. Yu, Y. Chen, C. Johnson, J. Sakakibara, H. Kiba, T. Sakurai, T. Tavares, R. Nusinow, D. A. Baxter, I. Schmutz, J. Brutnell, T. P and Kellogg, E. A. 2020. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nature Biotechnology 38: 1203-1210. Martienssen, R and McCombie, W. R. 2001. The first plant genome. Cell 105: 571-574. Michael, T. P. Bryant, D. Gutierrez, R. Borisjuk, N. Chu, P. Zhang, H. Xia, J. Zhou, J. Peng, H. El Baidouri, M. Hallers, B. T. Hastie, A. R. Liang, T. Acosta, K. Gilbert, S. McEntee, C. Jackson, S. A. Mockler, T. C. Zhang, W and Lam, E. 2017. Comprehensive definition of genome features in Spirodela polyrhiza by high-depth physical mapping and short-read DNA sequencing strategies. The Plant Journal 89: 617-635. Miles, B. N. Ivanov, A. P. Wilson, K. A. Doğan, F. Japrung, D and Edel, J. B. 2013. Single molecule sensing with solid-state nanopores: Novel materials, methods, and applications. Chemical Society Reviews 42: 15-28. Mitchell-Olds, T. 2001.
Arabidopsis thaliana and its wild relatives: A model system for ecology and evolution. Trends in Ecology and Evolution 16: 693-700. Mitros, T. Session, A. M. James, B. T. Wu, G. A. Belaffif, M. B. Clark, L. V. Shu, S. Dong, H. Barling, A. Holmes, J. R. Mattick, J. E. Bredeson, J. V. Liu, S. Farrar, K. Głowacka, K. Jeżowski, S. Barry, K. Chae, W. B. Juvik, J. A. Gifford, J. Oladeinde, A. Yamada, T. Grimwood, J. Putnam, N. H. Vega, J. De. Barth, S. Klaas, M. Hodkinson, T. Li, L. Jin, X. Peng, J. Yu, C. Y. Heo, K. Yoo, J. H. Ghimire, B. K. Donnison, I. S. Schmutz, J. Hudson, M. E. Sacks, E. J. Moose, S. P. Swaminathan, K and Rokhsar, D. S. 2020. Genome biology of the paleotetraploid perennial biomass crop Miscanthus
. Nature Communications 11: 5442. Naito, K. Wakatake, T. Shibata, T. F. Iseki, K. Shigenobu, S. Takahashi, Y. Ogiso-Tanaka, E. Muto, C. Teruya, K. Shiroma, A. Shimoji, M. Satou, K. Hirano, T. Nagano, A. J. Tomooka, N. Hasebe, M. Fukushima, K and Sakai, H. 2022. Genome sequence of 12 Vigna species as a knowledge base of stress tolerance and resistance. bioRxiv.
https://doi.org/10.1101/2022.03.28.486085. Nakamura, N. Hirakawa, H. Sato, S. Otagaki, S. Matsumoto, S. Tabata, S and Tanaka, Y. 2018. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Research 25: 113-121. Norn, S. Permin, H. Kruse, P. R and Kruse, E. 2009. From willow bark to acetylsalicylic acid. Dansk Medicinhistorisk Arbog 37: 79-98. Olsen, J. L. Rouzé, P. Verhelst, B. Lin, Y.-C. Bayer, T. Collen, J. Dattolo, E. Paoli, E. De. Dittami, S. Maumus, F. Michel, G. Kersting, A. Lauritano, C. Lohaus, R. Töpel, M. Tonon, T. Vanneste, K. Amirebrahimi, M. Brakel, J. Boström, C. Chovatia, M. Grimwood, J. Jenkins, J. W. Jueterbock, A. Mraz, A. Stam, W. T. Tice, H. Bornberg-Bauer, E. Green, P. J. Pearson, G. A. Procaccini, G. Duarte, C. M. Schmutz, J. Reusch, T. B. H and Van de Peer, Y. 2016. The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530: 331-335. Ossowski, S. Schneeberger, K. Clark, R. M. Lanz, C. Warthmann, N and Weigel, D. 2008. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Research 18: 2024-2033. Park, J. An, J.-H. Kim, Y. Kim, D. Yang, B-G and Kim, T. 2020a. Database of National Species List of Korea: The taxonomical systematics platform for managing scientific names of Korean native species. Journal of Species Research 9: 233-246.
Park, J. Kim, M. Xi, H. Park, S and Yun, Y. 2021a. Plant Genome Database Release 2.7: One tera base pairs plant genomes era. The 17th KOGO Winter Symposium. 2021 Feb 1–2. Seoul, Korea:
Park, J. Xi, H. Han, J. Lee, J. Kim, Y. Lee, J.-M. Son, J. Ahn, J. Jang, T. Choi, J and Park, J. 2020b. Prediction and identification of biochemical pathway of acteoside from whole genome sequences of Abeliophyllum distichum Nakai, cultivar Ok Hwang 1ho. Journal of Convergence for Information Technology 10: 76-91.
Park, J. Xi, H and Kim, Y. 2020c. The complete chloroplast genome of Arabidopsis thaliana isolated in Korea (Brassicaceae): An investigation of intraspecific variations of the chloroplast genome of Korean A. thaliana
. International Journal of Genomics 2020: 3236461. Pootakham, W. Naktang, C. Kongkachana, W. Sonthirod, C. Yoocha, T. Sangsrakru, D. Jomchai, N. U-thoomporn, S. Romyanon, K. Toojinda, T and Tangphatsornruang, S. 2021.
De novo chromosome-level assembly of the Centella asiatica genome. Genomics 113: 2221-2228. Ren, L. Guo, X. Liu, S. Yu, T. Guo, W. Wang, R. Ye, S. Lambertini, C. Brix, H and Eller, F. 2020. Intraspecific variation in Phragmites australis: Clinal adaption of functional traits and phenotypic plasticity vary with latitude of origin. Journal of Ecology 108: 2531-2543. Salojärvi, J. Smolander, O.-P. Nieminen, K. Rajaraman, S. Safronov, O. Safdari, P. Lamminmäki, A. Immanen, J. Lan, T. Tanskanen, J. Rastas, P. Amiryousefi, A. Jayaprakash, B. Kammonen, J. I. Hagqvist, R. Eswaran, G. Ahonen, V. H. Serra, J. A. Asiegbu, F. O. de Dios Barajas-Lopez, J. Blande, D. Blokhina, O. Blomster, T. Broholm, S. Brosché, M. Cui, F. Dardick, C. Ehonen, S. E. Elomaa, P. Escamez, S. Fagerstedt, K. V. Fujii, H. Gauthier, A. Gollan, PJ. Halimaa, P. Heino, PI. Himanen, K. Hollender, C. Kangasjärvi, S. Kauppinen, L. Kelleher, CT. Kontunen-Soppela, S. Koskinen, JP. Kovalchuk, A. Kärenlampi, SO. Kärkönen, AK. Lim, K-J. Leppälä, J. Macpherson, L. Mikola, J. Mouhu, K. Mähönen, AP. Niinemets, Ü. Oksanen, E. Overmyer, K. Palva, ET. Pazouki, L. Pennanen, V. Puhakainen, T. Poczai, P. Possen, BJHM. Punkkinen, M. Rahikainen, MM. Rousi, M. Ruonala, R. van der Schoot, C. Shapiguzov, A. Sierla, M. Sipilä, TP. Sutela, S. Teeri, TH. Tervahauta, Arja. I. Vaattovaara, A. Vahala, J. Vetchinnikova, L. Welling, A. Wrzaczek, M. Xu, E. Paulin, LG. Schulman, AH. Lascoux, M. Albert, VA. Auvinen, P. Helariutta, Y and Kangasjärvi, J. 2017. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nature Genetics 49: 904-912. Schmitz, R. J. Schultz, M. D. Urich, M. A. Nery, J. R. Pelizzola, M. Libiger, O. Alix, A. McCosh, RB. Chen, H. Schork, NJ and Ecker, JR. 2013. Patterns of population epigenomic diversity. Nature 495: 193-198. Schnable, P. S. Ware, D. Fulton, R. S. Stein, J. C. Wei, F. Pasternak, S. Liang, C. Zhang, J. Fulton, L. Graves, T. A. Minx, P. Reily, A. D. Courtney, L. Kruchowski, S. S. Tomlinson, C. Strong, C. Delehaunty, K. Fronick, C. Courtney, B. Rock, S. M. Belter, E. Du, F. Kim, K. Abbott, R. M. Cotton, M. Levy, A. Marchetto, P. Ochoa, K. Jackson, S. M. Gillam, B. Chen, W. Yan, L. Higginbotham, J. Cardenas, M. Waligorski, J. Applebaum, E. Phelps, L. Falcone, J. Kanchi, K. Thane, T. Scimone, A. Thane, N. Henke, J. Wang, T. Ruppert, J. Shah, N. Rotter, K. Hodges, J. Ingenthron, E. Cordes, M. Kohlberg, S. Sgro, J. Delgado, B. Mead, K. Chinwalla, A. Leonard, S. Crouse, K. Collura, K. Kudrna, D. Currie, J. He, R. Angelova, A. Rajasekar, S. Mueller, T. Lomeli, R. Scara, G. Ko, A. Delaney, K. Wissotski, M. Lopez, G. Campos, D. Braidotti, M. Ashley, E. Golser, W. Kim, H. Lee, S. Lin, J. Dujmic, Z. Kim, W. Talag, J. Zuccolo, A. Fan, C. Sebastian, A. Kramer, M. Spiegel, L. Nascimento, L. Zutavern, T. Miller, B. Ambroise, C. Muller, S. Spooner, W. Narechania, A. Ren, L. Wei, S. Kumari, S. Faga, B. Levy, M. J. McMahan, L. Van Buren, P. Vaughn, M. W. Ying, K. Yeh, C.-T. Emrich, S. J. Jia, Y. Kalyanaraman, A. Hsia, A.-P. Barbazuk, W. B. Baucom, R. S. Brutnell, T. P. Carpita, N. C. Chaparro, C. Chia, J.-M. Deragon, J.-M. Estill, J. C. Fu, Y. Jeddeloh, J. A. Han, Y. Lee, H. Li, P. Lisch, D. R. Liu, S. Liu, Z. Nagel, D. H. McCann, M. C. SanMiguel, P. Myers, A. M. Nettleton, D. Nguyen, J. Penning, B. W. Ponnala, L. Schneider, K. L. Schwartz, D. C. Sharma, A. Soderlund, C. Springer, N. M. Sun, Q. Wang, H. Waterman, M. Westerman, R. Wolfgruber, T. K. Yang, L. Yu, Y. Zhang, L. Zhou, S. Zhu, Q. Bennetzen, J. L. Dawe, R. K. Jiang, J. Jiang, N. Presting, G. G. Wessler, S. R. Aluru, S. Martienssen, R. A. Clifton, S. W. McCombie, W. R. Wing, R. A and Wilson, R. K. 2009. The B73 maize genome: Complexity, diversity, and dynamics. Science 326: 1112-1115. Shen, L.-Y. Luo, H. Wang, X-L. Wang, X-M. Qiu, X-J. Liu, H. Zhou, S-S. Jia, K-H. Nie, S. Bao, Y-T. Zhang, R-G. Yun, Q-Z. Chai, Y-H. Lu, J-Y. Li, Y. Zhao, S-W. Mao, J-F. Jia, S-G and Mao, Y-M. 2021. Chromosome-scale genome assembly for Chinese sour jujube and insights into its genome evolution and domestication signature. Frontiers in Plant Science 12: 773090. Shen, Q. Zhang, L. Liao, Z. Wang, S. Yan, T. Shi, P. Liu, M. Fu, X. Pan, Q. Wang, Y. Lv, Z. Lu, X. Zhang, F. Jiang, W. Ma, Y. Chen, M. Hao, X. Li, L. Tang, Y. Lv, G. Zhou, Y. Sun, X. Brodelius, P. E. Rose, J. K.C and Tang, K. 2018. The genome of Artemisia annua provides insight into the evolution of Asteraceae family and artemisinin biosynthesis. Molecular Plant 11: 776-788. Shirasawa, K. Itai, A and Isobe, S. 2021a. Chromosome-scale genome assembly of Japanese pear ( Pyrus pyrifolia) variety ‘Nijisseiki’. DNA Research 28: dsab001. Shirasawa, K. Kosugi, S. Sasaki, K. Ghelfi, A. Okazaki, K. Toyoda, A. Hirakawa, H and Isobe, S. 2021b. Genome features of common vetch ( Vicia sativa) in natural habitats. Plant Direct 5: e352. Shirasawa, K. Nishio, S. Terakami, S. Botta, R. Marinoni, D. T and Isobe, S. 2021c. Chromosome-level genome assembly of Japanese chestnut ( Castanea crenata Sieb. et Zucc.) reveals conserved chromosomal segments in woody rosids. DNA Research 28: dsab016. Shirasawa, K. Yakushiji, H. Nishimura, R. Morita, T. Jikumaru, S. Ikegami, H. Toyoda, A. Hirakawa, H and Isobe, S. 2020. The Ficus erecta genome aids Ceratocystis canker resistance breeding in common fig ( F. carica). The Plant Journal 102: 1313-1322. Slavov, G. T. DiFazio, S. P. Martin, J. Schackwitz, W. Muchero, W. Rodgers-Melnick, E. Lipphardt, M. F. Pennacchio, C. P. Hellsten, U. Pennacchio, L. A. Gunter, L. E. Ranjan, P. Vining, K. Pomraning, K. R. Wilhelm, L. J. Pellegrini, M. Mockler, T. C. Freitag, M. Geraldes, A. El-Kassaby, Y. A. Mansfield, S. D. Cronk, Q. C. B. Douglas, C. J. Strauss, S. H. Rokhsar, D and Tuskan, G. A. 2012. Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa
. New Phytologist 196: 713-725. Šmarda, P. Bureš, P. Horová, L. Leitch, IJ. Mucina, L. Pacini, E. Tichý, L. Grulich, V and Rotreklová, O. 2014. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proceedings of the National Academy of Sciences of the United States of America 111: E4096-E4102. Smith, A. M. Heisler, L. E. Onge, R. P. St. Farias-Hesson, E. Wallace, I. M. Bodeau, J. Harris, A. N. Perry, K. M. Giaever, G. Pourmand, N and Nislow, C. 2010. Highly-multiplexed barcode sequencing: An efficient method for parallel analysis of pooled samples. Nucleic Acids Research 38: e142. Sollars, E. S. A. Harper, A. L. Kelly, L. J. Sambles, C. M. Ramirez-Gonzalez, R. H. Swarbreck, D. Kaithakottil, G. Cooper, E. D. Uauy, C. Havlickova, L. Worswick, G. Studholme, D. J. Zohren, J. Salmon, D. L. Clavijo, B. J. Li, Y. He, Z. Fellgett, A. McKinney, L. V. Nielsen, L. R. Douglas, G. C. Kjær, E. D. Downie, J. A. Boshier, D. Lee, S. Clark, J. Grant, M. Bancroft, I. Caccamo, M and Buggs, R. J. A. 2017. Genome sequence and genetic diversity of European ash trees. Nature 541: 212-216. Song, X. Li, Y. Cao, X and Qi, Y. 2019. MicroRNAs and their regulatory roles in plant–environment interactions. Annual Review of Plant Biology 70: 489-525. Staton, M. Addo-Quaye, C. Cannon, N. Yu, J. Zhebentyayeva, T. Huff, M. Islam-Faridi, N. Fan, S. Georgi, LL. Nelson, CD. Bellis, E. Fitzsimmons, S. Henry, N. Drautz-Moses, D. Noorai, RE. Ficklin, S. Saski, C. Mandal, M. Wagner, TK. Zembower, N. Bodénès, C. Holliday, J. Westbrook, J. Lasky, J. Hebard, FV. Schuster, SC. Abbott, A. G and Carlson, J. E. 2020. A reference genome assembly and adaptive trait analysis of Castanea mollissima ‘Vanuxem,’ a source of resistance to chestnut blight in restoration breeding. Tree Genetics and Genomes 16: 57. Sun, G. Xu, Y. Liu, H. Sun, T. Zhang, J. Hettenhausen, C. Shen, G. Qi, J. Qin, Y. Li, J. Wang, L. Chang, W. Guo, Z. Baldwin, I. T and Wu, J. 2018. Large-scale gene losses underlie the genome evolution of parasitic plant Cuscuta australis
. Nature Communications 9: 2683. Tahir, J. Crowhurst, R. Deroles, S. Hilario, E. Deng, C. Schaffer, R. Lievre, L. Le. Brendolise, C. Chagne, D. Gardiner, S. E. Knaebel, M. Catanach, A. McCallum, J. Datson, P. Thomson, S. Brownfield, L. R. Nardozza, S and Pilkington, S. M. 2022. First chromosome-scale assembly and deep floral-bud transcriptome of a male kiwifruit. Frontiers in Genetics 13: 852161. Tan, Q. Li, S. Zhang, Y. Chen, M. Wen, B. Jiang, S. Chen, X. Fu, X. Li, D. Wu, H. Wang, Y. Xiao, W and Li, L. 2021. Chromosome- level genome assemblies of five Prunus species and genome-wide association studies for key agronomic traits in peach. Horticulture Research 8: 213. Tanaka, H. Hirakawa, H. Kosugi, S. Nakayama, S. Ono, A. Watanabe, A. Hashiguchi, M. Gondo, T. Ishigaki, G. Muguerza, M. Shimizu, K. Sawamura, N. Inoue, T. Shigeki, Y. Ohno, N. Tabata, S. Akashi, R and Sato, S. 2016. Sequencing and comparative analyses of the genomes of zoysiagrasses. DNA Research 23: 171-180. The 1001 Genomes Consortium. 2016. 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana
. Cell 166: 481-491. The Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana
. Nature 408: 796-815. Tuskan, GA. DiFazio, S. Jansson, S. Bohlmann, J. Grigoriev, I. Hellsten, U. Putnam, N. Ralph, S. Rombauts, S. Salamov, A. Schein, J. Sterck, L. Aerts, A. Bhalerao, RR. Bhalerao, RP. Blaudez, D. Boerjan, W. Brun, A. Brunner, A. Busov, V. Campbell, M. Carlson, J. Chalot, M. Chapman, J. Chen, G-L. Cooper, D. Coutinho, PM. Couturier, J. Covert, S. Cronk, Q. Cunningham, R. Davis, J. Degroeve, S. Déjardin, A. Depamphilis, C. Detter, J. Dirks, B. Dubchak, I. Duplessis, S. Ehlting, J. Ellis, B. Gendler, K. Goodstein, D. Gribskov, M. Grimwood, J. Groover, A. Gunter, L. Hamberger, B. Heinze, B. Helariutta, Y. Henrissat, B. Holligan, D. Holt, R. Huang, W. Islam-Faridi, N. Jones, S. Jones-Rhoades, M. Jorgensen, R. Joshi, C. Kangasjärvi, J. Karlsson, J. Kelleher, C. Kirkpatrick, R. Kirst, M. Kohler, A. Kalluri, U. Larimer, F. Leebens-Mack, J. Leplé, J-C. Locascio, P. Lou, Y. Lucas, S. Martin, F. Montanini, B. Napoli, C. Nelson, D. R. Nelson, C. Nieminen, K. Nilsson, O. Pereda, V. Peter, G. Philippe, R. Pilate, G. Poliakov, A. Razumovskaya, J. Richardson, P. Rinaldi, C. Ritland, K. Rouzé, P. Ryaboy, D. Schmutz, J. Schrader, J. Segerman, B. Shin, H. Siddiqui, A. Sterky, F. Terry, A. Tsai, C-J. Uberbacher, E. Unneberg, P. Vahala, J. Wall, K. Wessler, S. Yang, G. Yin, T. Douglas, C. Marra, M. Sandberg, G. Van de Peer, Y and Rokhsar, D. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313: 1596-1604. Valliyodan, B. Cannon, S. B. Bayer, P. E. Shu, S. Brown, A. V. Ren, L. Jenkins, J. Chung, C. Y.-L. Chan, T.-F. Daum, C. G. Plott, C. Hastie, A. Baruch, K. Barry, K. W. Huang, W. Patil, G. Varshney, R. K. Hu, H. Batley, J. Yuan, Y. Song, Q. Stupar, R. M. Goodstein, D. M. Stacey, G. Lam, H.-M. Jackson, S. A. Schmutz, J. Grimwood, J. Edwards, D and Nguyen, H. T. 2019. Construction and comparison of three reference-quality genome assemblies for soybean. The Plant Journal 100: 1066-1082. Van Hoeck, A. Horemans, N. Monsieurs, P. Cao, H. X. Vandenhove, H and Blust, R. 2015. The first draft genome of the aquatic model plant Lemna minor opens the route for future stress physiology research and biotechnological applications. Biotechnology for Biofuels 8: 188. Venter, J. C. Adams, M. D. Myers, E. W. Li, P. W. Mural, R. J. Sutton, G. G. Smith, H. O. Yandell, M. Evans, C. A. Holt, R. A. Gocayne, J. D. Amanatides, P. Ballew, R. M. Huson, D. H. Wortman, J. R. Zhang, Q. Kodira, C. D. Zheng, X. H. Chen, L. Skupski, M. Subramanian, G. Thomas, P. D. Zhang, J. Gabor Miklos, G. L. Nelson, C. Broder, S. Clark, A. G. Nadeau, J. McKusick, V. A. Zinder, N. Levine, A. J. Roberts, R. J. Simon, M. Slayman, C. Hunkapiller, M. Bolanos, R. Delcher, A. Dew, I. Fasulo, D. Flanigan, M. Florea, L. Halpern, A. Hannen-halli, S. Kravitz, S. Levy, S. Mobarry, C. Reinert, K. Remington, K. Abu-Threideh, J. Beasley, E. Biddick, K. Bonazzi, V. Brandon, R. Cargill, M. Chandramouliswaran, I. Charlab, R. Chaturvedi, K. Deng, Z. Di Francesco, V. Dunn, P. Eilbeck, K. Evangelista, C. Gabrielian, A. E. Gan, W. Ge, W. Gong, F. Gu, Z. Guan, P. Heiman, T. J. Higgins, M. E. Ji, R. R. Ke, Z. Ketchum, K. A. Lai, Z. Lei, Y. Li, Z. Li, J. Liang, Y. Lin, X. Lu, F. Merkulov, G. V. Milshina, N. Moore, H. M. Naik, A. K. Narayan, V. A. Neelam, B. Nusskern, D. Rusch, D. B. Salzberg, S. Shao, W. Shue, B. Sun, J. Wang, Z. Wang, A. Wang, X. Wang, J. Wei, M. Wides, R. Xiao, C. Yan, C. Yao, A. Ye, J. Zhan, M. Zhang, W. Zhang, H. Zhao, Q. Zheng, L. Zhong, F. Zhong, W. Zhu, S. Zhao, S. Gilbert, D. Baumhueter, S. Spier, G. Carter, C. Cravchik, A. Woodage, T. Ali, F. An, H. Awe, A. Baldwin, D. Baden, H. Barnstead, M. Barrow, I. Beeson, K. Busam, D. Carver, A. Center, A. Cheng, M. L. Curry, L. Danaher, S. Davenport, L. Desilets, R. Dietz, S. Dodson, K. Doup, L. Ferriera, S. Garg, N. Gluecksmann, A. Hart, B. Haynes, J. Haynes, C. Heiner, C. Hladun, S. Hostin, D. Houck, J. Howland, T. Ibegwam, C. Johnson, J. Kalush, F. Kline, L. Koduru, S. Love, A. Mann, F. May, D. McCawley, S. McIntosh, T. McMullen, I. Moy, M. Moy, L. Murphy, B. Nelson, K. Pfannkoch, C. Pratts, E. Puri, V. Qureshi, H. Reardon, M. Rodriguez, R. Rogers, Y. H. Romblad, D. Ruhfel, B. Scott, R. Sitter, C. Smallwood, M. Stewart, E. Strong, R. Suh, E. Thomas, R. Tint, N. N. Tse, S. Vech, C. Wang, G. Wetter, J. Williams, S. Williams, M. Windsor, S. Winn-Deen, E. Wolfe, K. Zaveri, J. Zaveri, K. Abril, J. F. Guigó, R. Campbell, M. J. Sjolander, K. V. Karlak, B. Kejariwal, A. Mi, H. Lazareva, B. Hatton, T. Narechania, A. Diemer, K. Muruganujan, A. Guo, N. Sato, S. Bafna, V. Istrail, S. Lippert, R. Schwartz, R. Walenz, B. Yooseph, S. Allen, D. Basu, A. Baxendale, J. Blick, L. Caminha, M. Carnes-Stine, J. Caulk, P. Chiang, Y. H. Coyne, M. Dahlke, C. Deslattes Mays, A. Dombroski, M. Donnelly, M. Ely, D. Esparham, S. Fosler, C. Gire, H. Glanowski, S. Glasser, K. Glodek, A. Gorokhov, M. Graham, K. Gropman, B. Harris, M. Heil, J. Henderson, S. Hoover, J. Jennings, D. Jordan, C. Jordan, J. Kasha, J. Kagan, L. Kraft, C. Levitsky, A. Lewis, M. Liu, X. Lopez, J. Ma, D. Majoros, W. McDaniel, J. Murphy, S. Newman, M. Nguyen, T. Nguyen, N. Nodell, M. Pan, S. Peck, J. Peterson, M. Rowe, W. Sanders, R. Scott, J. Simpson, M. Smith, T. Sprague, A. Stockwell, T. Turner, R. Venter, E. Wang, M. Wen, M. Wu, D. Wu, M. Xia, A. Zandieh, A and Zhu, X. 2001. The sequence of the human genome. Science 291: 1304-1351. Wang, J. Wang, W. Li, R. Li, Y. Tian, G. Goodman, L. Fan, W. Zhang, J. Li, J. Zhang, J. Guo, Y. Feng, B. Li, H. Lu, Y. Fang, X. Liang, H. Du, Z. Li, D. Zhao, Y. Hu, Y. Yang, Z. Zheng, H. Hellmann, I. Inouye, M. Pool, J. Yi, X. Zhao, J. Duan, J. Zhou, Y. Qin, J. Ma, L. Li, G. Yang, Z. Zhang, G. Yang, B. Yu, C. Liang, F. Li, W. Li, S. Li, D. Ni, P. Ruan, J. Li, Q. Zhu, H. Liu, D. Lu, Z. Li, S. Guo, G. Zhang, J. Ye, J. Fang, L. Hao, Q. Chen, Q. Liang, Y. Su, Y. San, A. Ping, C. Yang, S. Chen, F. Li, L. Zhou, K. Zheng, H. Ren, Y. Yang, L. Gao, Y. Yang, G. Li, Z. Feng, X. Kristiansen, K. Wong, G. K.-S. Nielsen, R. Durbin, R. Bolund, L. Zhang, X. Li, S. Yang, H and Wang, J. 2008. The diploid genome sequence of an Asian individual. Nature 456: 60-65. Wang, W. Haberer, G. Gundlach, H. Gläßer, C. Nussbaumer, T. Luo, M. Lomsadze, A. Borodovsky, M. Kerstetter, R. Shanklin, J. Byrant, D. W. Mockler, T. C. Appenroth, K. J. Grimwood, J. Jenkins, J. Chow, J. Choi, C. Adam, C. Cao, X.-H. Fuchs, J. Schubert, I. Rokhsar, D. Schmutz, J. Michael, T. P. Mayer, K. F. X and Messing, J. 2014. The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nature Communications 5: 3311. Wang, Y. Li, F. He, Q. Bao, Z. Zeng, Z. An, D. Zhang, T. Yan, L. Wang, H. Zhu, S and Liu, T. 2021a. Genomic analyses provide comprehensive insights into the domestication of bast fiber crop ramie ( Boehmeria nivea). The Plant Journal 107: 787-800. Wang, Y. Xin, H. Fan, P. Zhang, J. Liu, Y. Dong, Y. Wang, Z. Yang, Y. Zhang, Q. Ming, R. Zhong, G-Y. Li, S and Liang, Z. 2021b. The genome of Shanputao ( Vitis amurensis) provides a new insight into cold tolerance of grapevine. The Plant Journal 105: 1495-1506. Weber, J. L and Myers, E. W. 1997. Human whole-genome shotgun sequencing. Genome Research 7: 401-409. Williams, L. J. S. Tabbaa, DG. Li, N. Berlin, AM. Shea, TP. MacCallum, I. Lawrence, MS. Drier, Y. Getz, G. Young, SK. Jaffe, DB. Nusbaum, C and Gnirke, A. 2012. Paired-end sequencing of Fosmid libraries by Illumina. Genome Research 22: 2241-2249. Wu, H. Yao, D. Chen, Y. Yang, W. Zhao, W. Gao, H and Tong, C. 2020.
De novo genome assembly of Populus simonii further supports that Populus simonii and Populus trichocarpa belong to different sections. G3: Genes, Genomes, Genetics 10: 455-466. Xie, M. Chung, C. Y.-L. Li, M.-W. Wong, F.-L. Wang, X. Liu, A. Wang, Z. Leung, A. K.-Y. Wong, T.-H. Tong, S.-W. Xiao, Z. Fan, K. Ng, M.-S. Qi, X. Yang, L. Deng, T. He, L. Chen, L. Fu, A. Ding, Q. He, J. Chung, G. Isobe, S. Tanabata, T. Valliyodan, B. Nguyen, H. T. Cannon, S. B. Foyer, C. H. Chan, T.-F and Lam, H.-M. 2019. A reference-grade wild soybean genome. Nature Communications 10: 1216. Xing, Y. Liu, Y. Zhang, Q. Nie, X. Sun, Y. Zhang, Z. Li, H. Fang, K. Wang, G. Huang, H. isseling, T. Cao, Q and Qin, L. 2019. Hybrid de novo genome assembly of Chinese chestnut ( Castanea mollissima). GigaScience 8: giz112. Xu, J. Chu, Y. Liao, B. Xiao, S. Yin, Q. Bai, R. Su, H. Dong, L. Li, X. Qian, J. Zhang, J. Zhang, Y. Zhang, X. Wu, M. Zhang, J. Li, G. Zhang, L. Chang, Z. Zhang, Y. Jia, Z. Liu, Z. Afreh, D. Nahurira, R. Zhang, L. Cheng, R. Zhu, Y. Zhu, G. Rao, W. Zhou, C. Qiao, L. Huang, Z. Cheng, Y.-C and Chen, S. 2017.
Panax ginseng genome examination for ginsenoside biosynthesis. GigaScience 6: gix093. Yin, M. Zhang, S. Du, X. Mateo, R. G. Guo, W. Li, A. Wang, Z. Wu, S. Chen, J. Liu, J and Ren, G. 2021. Genomic analysis of Medicago ruthenica provides insights into its tolerance to abiotic stress and demographic history. Molecular Ecology Resources 21: 1641-1657. Yuan, Y. Jin, X. Liu, J. Zhao, X. Zhou, J. Wang, X. Wang, D. Lai, C. Xu, W. Huang, J. Zha, L. Liu, D. Ma, X. Wang, L. Zhou, M. Jiang, Z. Meng, H. Peng, H. Liang, Y. Li, R. Jiang, C. Zhao, Y. Nan, T. Jin, Y. Zhan, Z. Yang, J. Jiang, W and Huang, L. 2018. The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nature Communications 9: 1615. Zhang, H. Hall, N. Goertzen, LR. Bi, B. Chen, CY. Peatman, E. Lowe, EK. Patel, J and McElroy, J. S. 2019. Development of a goosegrass ( Eleusine indica) draft genome and application to weed science research. Pest Management Science 75: 2776-2784. Zhang, H.-B and Wu, C. 2001. BAC as tools for genome sequencing. Plant Physiology and Biochemistry 39: 195-209. Zimin, A. Stevens, KA. Crepeau, MW. Holtz-Morris, A. Koriabine, M. Marçais, G. Puiu, D. Roberts, M. Wegrzyn, JL. de Jong, PJ. Neale, DB. Salzberg, SL. Yorke, J. A and Langley, C. H. 2014. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics 196: 875-890. Zou, Y.-P. Hou, X.-H. Wu, Q. Chen, J.-F. Li, Z.-W. Han, T.-S. Niu, X.- M. Yang, L. Xu, Y.-C. Zhang, J. Zhang, F.-M. Tan, D. Tian, Z. Gu, H and Guo, Y.-L. 2017. Adaptation of Arabidopsis thaliana to the Yangtze River basin. Genome Biology 18: 239. Züst, T. Strickler, SR. Powell, AF. Mabry, ME. An, H. Mirzaei, M. York, T. Holland, CK. Kumar, P. Erb, M. Petschenka, G. Gómez, J-M. Perfectti, F. Müller, C. Pires, JC. Mueller, L. A and Jander, G. 2020. Independent evolution of ancestral and novel defenses in a genus of toxic plants ( Erysimum, Brassicaceae). Elife 9: e51712.
|
|











