Comparative analyses of Agrimonia complete chloroplast genomes with newly assembled chloroplast genomes of A. coreana and A. nipponica (Rosaceae)
Article information

Abstract
The genus Agrimonia L. (1753) is a small group consisting of 19 species and three varieties. We completed two Agrimonia chloroplast genomes, Agrimonia coreana Nakai and Agrimonia nipponica Koidz, collected in Korea and conducted comprehensive analyses of six Agrimonia chloroplast genomes. Two of the Agrimonia chloroplasts genomes have a typical quadripartite structure with lengths ranging from 155,161 bp to 151,362 bp, similar to the remaining Agrimonia chloroplast genomes. High nucleotide diversity was detected in the ycf1 gene, which can serve as a molecular marker. Intraspecific variations of four A. pilosa chloroplast genomes and interspecific variations of three Agrimonia species revealed that KY419942 is distinct to the other three A. pilosa chloroplast genomes. A phylogenetic analysis demonstrated that KY419942 was clustered with A. nipponica, requiring further analyses to understand Agrimonia species in East Asia. A comparison of simple sequence repeats identified from the six Agrimonia chloroplast genomes suggests potential molecular markers to distinguish species or populations of the same species. Our results define the phylogenetic relationship of three Agrimonia species and provide insight into the intraspecific features of Agrimonia chloroplast genomes with which to gain a better understanding of the genetic structure of Agrimonia species.
INTRODUCTION
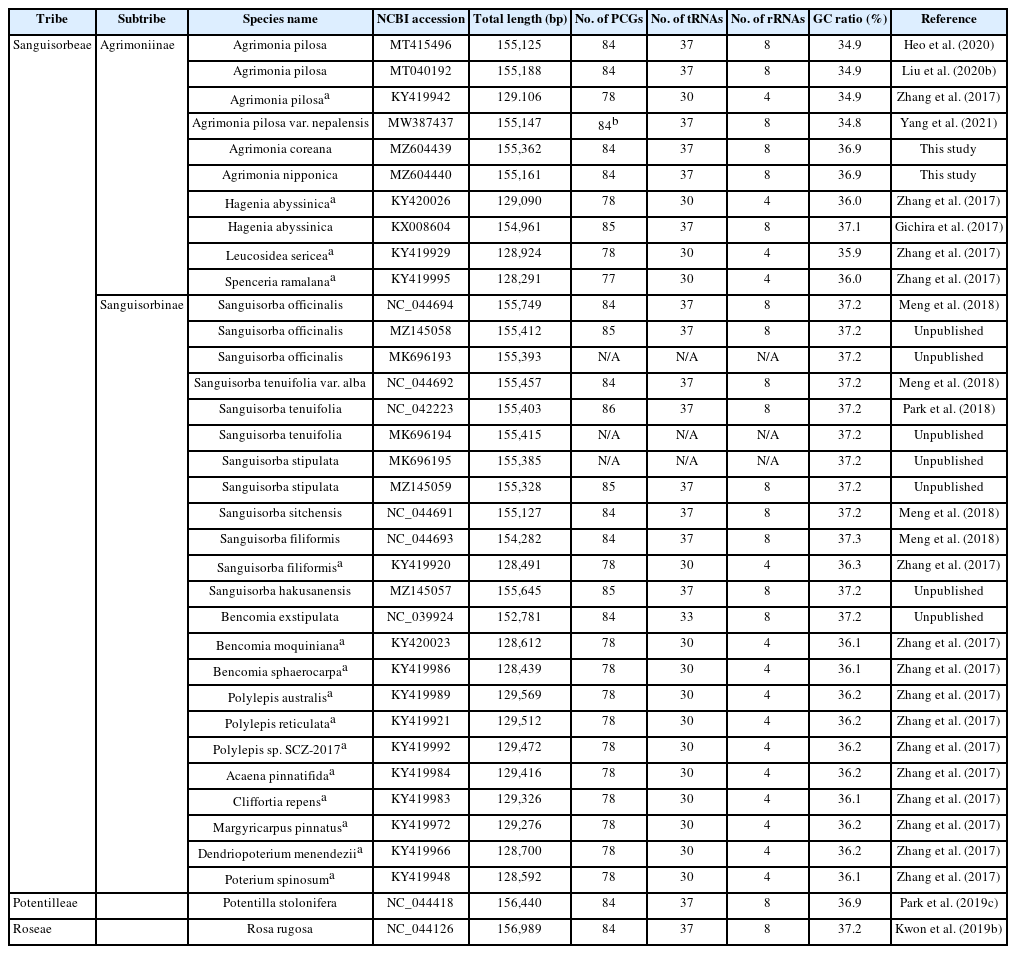
The subtribe Agrimoniinae (Rosaceae, Rosoideae), one of two members of the tribe Sanguisorbeae, displays polyploidy and morphological variations, both of which are good sources of information when attempting to understand the corresponding biogeographical patterns and endemism (Chung, 2008). This subtribe consists of the five genera, Agrimonia (19 species in Eurasia and South America), Hagenia (one species in east Africa), Leucosidea (one species in southern Africa), Spenceria (one species in central Asia), and Aremonia (one species in southern Europe) (Potter et al., 2007; Chung 2008), with all genera except Agrimonia being monotypic. Given this biased distribution of species, partial or complete chloroplast genomes for four out of the five genera except Aremonia have been determined thus far: two complete and one partial determination for Agrimonia pilosa Ledeb. (Zhang et al., 2017; Heo et al., 2020; Liu et al., 2020b), one complete determination for Agrimonia pilosa var. nepalensis (D. Don) Nakai (Yang et al., 2021), one complete and one partial determination for Hagenia abyssinica Willd (Gichira et al., 2017; Zhang et al., 2017), one partial determination for Leucosidea sericea Eckl. & Zeyh. (Zhang et al., 2017), and one partial determination for Spenceria ramalana Trimen (Zhang et al., 2017).
Agrimonia L. (1753), consisting of 19 species and three varieties, is characterized by two major morphological characteristics: interrupted pinnately compound leaves and bristly indumentum, within Rosaceae (Chung, 2008). Agrimonia species are usually distributed in temperate regions, including North America, Central America, the West Indies, southern South America, Europe, Asia, and southern Africa (Hutchinson, 1964; Skalický, 1973; Robertson, 1974; Murata and Umemoto, 1983; Chung and Kim, 2000; Kalkman, 2004; Chung, 2008). The base chromosome number is x = 7 (Darlington and Wylie, 1955) and the polyploidy levels of Agrimonia species range from tetraploid (2n = 4x = 28) to octaploid (2n = 8x = 56) (Chung, 2008). Various natural hybrids observed in Agrimonia hinder a clear delimitation of its species boundary (Wallroth, 1842; Skalický, 1962; Chung, 2008), indicating that alternative methods, such as the use of molecular markers, are required to understand the species boundary.
The Agrimonia pilosa complex is an excellent case for investigating molecular variation at the genomic level. The A. pilosa complex, with three morphologically similar species A. pilosa, A. coreana Nakai, and A. nipponica Koidz, is characterized by having relatively small fruits with spreading, erect, or connivent prickles (Chung and Kim, 2000; Chung, 2008). All of these species are distributed in East Asia with extensions to Eastern Europe. An examination of the intraspecific variations of chloroplast genomes together with their morphological characteristics would provide fundamental information about the levels and patterns of variations within and among closely related species, especially considering that data pertaining to four chloroplast genomes of A. pilosa are available in China and Korea (Zhang et al., 2017; Heo et al., 2020; Liu et al., 2020b). In addition, plants of the A. pilosa complex have been used to treat boils, eczema, taeniasis, abdominal pain, sore throat, headaches, and heat stroke in traditional medicine in China, Nepal, and Korea (World Health Organization, 1998). Careful and thorough investigations have also found that A. pilosa extracts have various useful effects, among them antiviral (Shin et al., 2010), whitening (Kim et al., 2011a; Kim et al., 2011b), antinociceptive (Park et al., 2012), antioxidant (Seo et al., 2008; Kim et al., 2011b; Chen and Kang, 2014), anticancer (Seo et al., 2008), anti-aging (Yoon et al., 2012), and anti-inflammatory (Jung et al., 2010) effects, suggesting the potential value of investigating the genetic background of Agrimonia species in Korea.
Owing to next-generation sequencing and third-generation sequencing technologies (Zhou et al., 2010; Roberts et al., 2013; Van Dijk et al., 2014; Deamer et al., 2016; Goodwin et al., 2016), nearly ten thousand chloroplast genomes have been sequenced thus far (Park et al., 2021d). These chloroplast genomes have been used for phylogenetic or phylogenomic analyses (Hassemer et al., 2019; Alzahrani et al., 2020; Liang et al., 2020; Chang et al., 2021), to identify the phylogenetic positions of new species candidates (Kim et al., 2019f; Oh et al., 2019a; Park et al., 2021b), to investigate intraspecific variations on chloroplast genomes (Li et al., 2018; Bum et al., 2020; Baek et al., 2021), and to develop useful molecular markers (Li et al., 2020a; Wang et al., 2020). Complete chloroplast genomes can be utilized to understand the relationships between morphological features and their phylogenetic positions. Examples of this possibility include Chenopodium album L. (Park et al., 2021d), Suaeda japonica Makino (Kim et al., 2020), the Allium genus (Xie et al., 2019), and the Fagopyrum genus (Wang et al., 2017). Due to the morphological complexity and the presence of intermediate forms in the A. pilosa complex, additional chloroplast genomes of A. coreana, and A. nipponica beyond the three chloroplast genomes of A. pilosa already reported (Zhang et al., 2017; Heo et al., 2020; Liu et al., 2020b) can be helpful to understand the relationships, morphological diversity and phylogenetic positions of this complex.
Here, we complete the two chloroplast genomes of A. coreana and A. nipponica and conduct comparative analyses of the six Agrimonia chloroplast genomes available to date. Our comprehensive analyses of nucleotide diversity, intraspecific and interspecific variations, and phylogeneticity in these genomes and comparisons of simple sequence repeats (SSRs) demonstrate that two groups of A. pilosa, one a sister to A. nipponica and the other a sister to A. coreana, reflect that A. coreana, A. nipponica, and A. pilosa have complex evolutionary histories. Also presented are the chromosome configurations of the three species. Further analyses of additional chloroplast genomes and the continuing whole genome sequencing of other Agrimonia species will be required to understand the precise evolutionary history of the Agrimonia genus.
MATERIALS AND METHODS
DNA extraction of two Agrimonia samples isolated in Korea
Two Agrimonia species, A. coreana and A. nipponica, were collected on the Korean Peninsula (Table 1). Both are native Korean plant species (Park et al., 2020a) collectable without special permission in Korea. The sampling process for Agrimonia was conducted while also adhering to all local, national, and international guidelines and laws. All vouchers of the two Agrimonia samples were deposited into the InfoBoss Cyber Herbarium (IN) (Table 1). Their total DNA was extracted from fresh leaves of the two samples using a DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany).
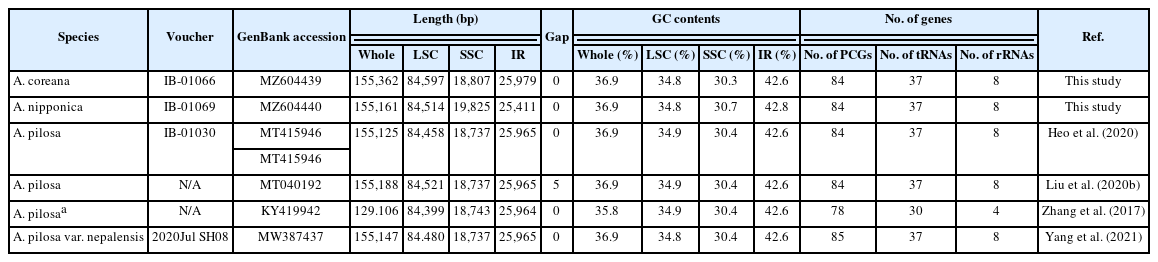
List of six Agrimonia chloroplast genomes analyzed in this study.
Genome sequencing and de novo assembly of the two Agrimonia chloroplast genomes
Genome sequencing was performed with the NovaSeq 6000 system at Macrogen Inc. in Korea using the DNA extracted from the two Agrimonia species. De novo assembly was then done, with confirmation accomplished with Velvet v1.2.10 (Zerbino and Birney, 2008) after filtering raw reads using Trimmomatic v0.33 (Bolger et al., 2014). After obtaining the first draft of the chloroplast genome sequences, gaps were filled with GapCloser v1.12 (Zhao et al., 2011) and all bases from the assembled sequences were confirmed by checking each base in the alignment (tview mode in SAMtools v1.9) (Li et al., 2009) against the assembled chloroplast genome generated with BWA v0.7.17 (Li, 2013). All of these processes were conducted under the environment of Genome Information System (GeIS; http://geis.infoboss.co.kr/), which has been utilized in a range of studies (Kim et al., 2021b, 2021c; Park et al., 2021c, 2021h), including work on plant organelle genomes (Suh et al., 2021; Park et al., 2022a, 2022c; Yoo et al., 2023).
Chloroplast genome annotation
Geneious Prime 2020.2.4 (Biomatters Ltd., Auckland, New Zealand) was used for genome annotation of the two Agrimonia species based on the A. pilosa chloroplast genome (GenBank accession number: MT415946) (Heo et al., 2020) by transferring annotations while correcting exceptional cases, including missing start or stop codons. tRNA was predicted and confirmed based on predictions by tRNAscan-SE v2.0.6 (Lowe and Chan, 2016). Circular maps of the two Agrimonia chloroplasts were drawn using the OGDRAW v1.3.1 (Greiner et al., 2019).
Identification of sequence variations from complete chloroplast genomes
Single nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs) were identified by means of a pair-wise alignment of the two selected chloroplast genomes done using MAFFT v7.450 (Katoh and Standley, 2013). When the numbers of INDELs were calculated, continuous INDEL bases were considered as one INDEL. When partial chloroplast genomes without one of the inverted repeat (IR) regions were used to identify sequence variations, SNPs and INDELs located in the IR regions were counted once more with the assumption that the IR region missing in the partial chloroplast genome is identical to the IR region in the partial chloroplast genomes. These methods have been utilized in previous studies (Kim et al., 2021a, 2021e; Park et al., 2021g).
Identification of SSRs
SSRs were identified on the chloroplast genome sequence using the pipeline of the SSR database (SSRDB; http://ssrdb.infoboss.co.kr/; Park et al., in preparation), as utilized in previous studies (Hong et al., 2022). Based on the conventional definition of an SSR on a chloroplast genome, monoSSR (1 bp) to hexaSSR (6 bp), the total length of the SSRs on the chloroplast genome exceeds 10 bp. Owing to the different criteria pertaining to SSRs on chloroplast genomes, we adopted the criteria used with the organelle genomes of Dysphania species (Kim et al., 2019d), Arabidopsis thaliana L. (Park et al., 2020c), Chenopodium album (Park et al., 2021d), Diarthron linifolium Turcz. (Kim et al., 2021d), Campsis grandiflora (Park and Xi 2022), and Rosa rugosa Thunb. (Park et al., 2020d), as follows; the monoSSR (unit sequence length of 1 bp) to hexaSSR (6 bp) are used as normal SSRs, and the heptaSSR (7 bp) to decaSSR (10 bp) are defined as the extended SSRs. Among the normal SSRs, pentaSSRs, and hexaSSRs with a unit sequence repeat number of 2 are classified as potential SSRs. Regional classifications of the chloroplast genome were conducted using the method described in the section above.
Comparison of SSRs identified from the six Agrimonia chloroplast genomes
SSRs identified from ten Agrimonia chloroplast genomes were compared based on their flanking sequences under the SSRDB environment (http://ssrdb.infoboss.co.kr/) (Park et al., in preparation). The pipeline of the SSR comparison implemented in the SSRDB relies on various organelle genome studies (Park et al., 2021i, 2022b; Kim et al., 2023) and was used with the following conditions: a cut-off e-value of 1e-10 and a maximum flanking sequence for the comparison of 60 bp.
Nucleotide diversity analysis
Nucleotide diversity was calculated using the method proposed by Nei and Li (Nei and Li, 1979) based on the multiple sequence alignment of Agrimonia chloroplast genomes using a Perl script used in previous studies (Lee et al., 2020; Choi et al., 2021). The window size was set to 500 bp and the step size was 200 bp when using the sliding-window method. The genomic coordination of each window was compared to the gene annotation of the chloroplast genome under the GenomeArchive (http://www.genomearchive.net/) environment for further analyses.
Construction of phylogenetic trees
Thirty-three whole chloroplast genomes of tribe Sanguisorbeae and two outgroup species, Rosa and Potentilla (Table 2), were aligned by MAFFT v7.450 (Katoh and Standley, 2013) and the alignment quality was checked manually. The maximum likelihood (ML) tree was reconstructed in IQ-TREE v1.6.6 (Nguyen et al., 2015). In the ML analysis, a heuristic search was used with nearest-neighbor interchange branch swapping, the GTR + F + R4 model, and uniform rates among sites. All other options used default settings. Bootstrap analyses with 1,000 pseudoreplicates were conducted with the same options. The posterior probability of each node was estimated by means of Bayesian inference (BI) using the MrBayes v3.2.6 (Huelsenbeck and Ronquist, 2001) plug-in implemented in Geneious Prime 2020.2.4 (Biomatters Ltd.). The HKY85 model with gamma rates was used as a molecular model. A Markov-chain Monte Carlo (MCMC) algorithm was employed for 1,100,000 generations, sampling trees every 200 generations, with four chains running simultaneously. Trees from the first 100,000 generations were discarded as burn-in.
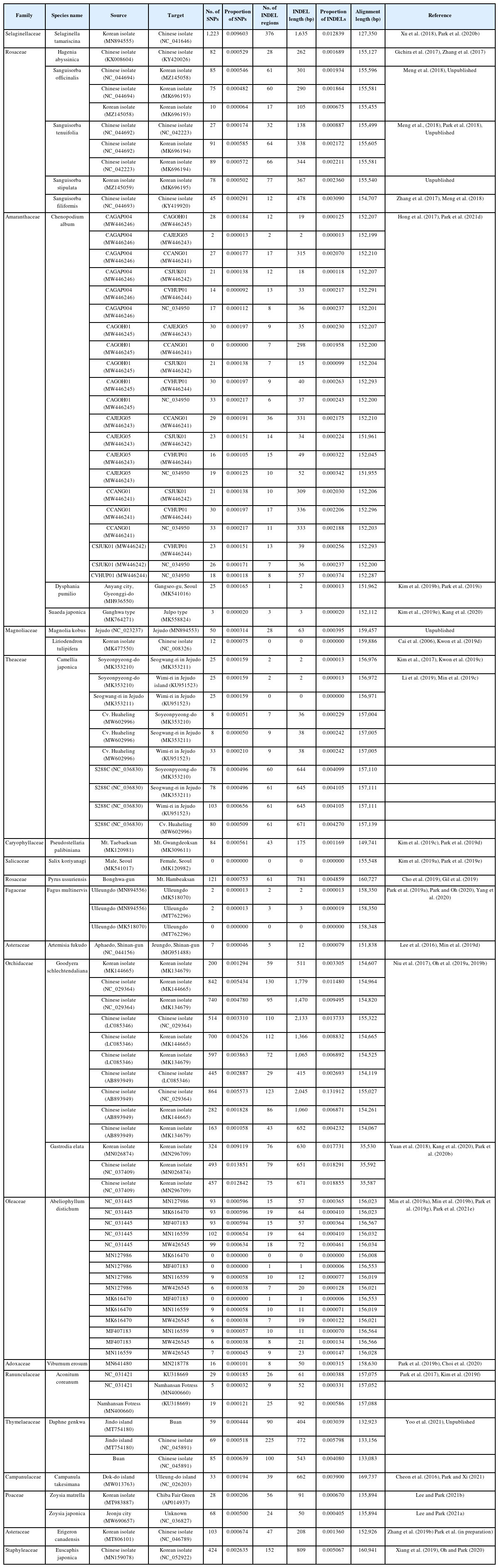
List of intraspecific variations identified from various plant species isolated from Korea and China using pair-wise comparison method.
RESULTS AND DISCUSSION
Two complete Agrimonia chloroplast genomes, A. coreana and A. nipponica
Two Agrimonia species, A. coreana and A. nipponica, were sampled on the Korean Peninsula (Table 1) (Chung 2017; Park et al., 2020e). Chloroplast genomes of the two Agrimonia species were successfully completed, with corresponding coverage rates of 133.88x and 150.31x. It was thus found that the A. coreana chloroplast genome was 151,362 bp long (Fig. 1A), the longest among the six Agrimonia chloroplast genomes, while that of A. nipponica was 155,161 bp long (Fig. 1B), similar to the other Agrimonia chloroplast genomes (Table 1). A small single-copy (SSC) region of A. nipponica was the longest (19,825 bp) among the six Agrimonia chloroplast genomes, while the IR region of A. nipponica was the shortest (25,411 bp). A large single-copy (LSC) region of A. coreana was longest (84,597 bp) (Table 1). The two Agrimonia chloroplast genome sequences sequenced in this study can be accessed via accession numbers MZ604439 and MZ604440 in the NCBI GenBank.
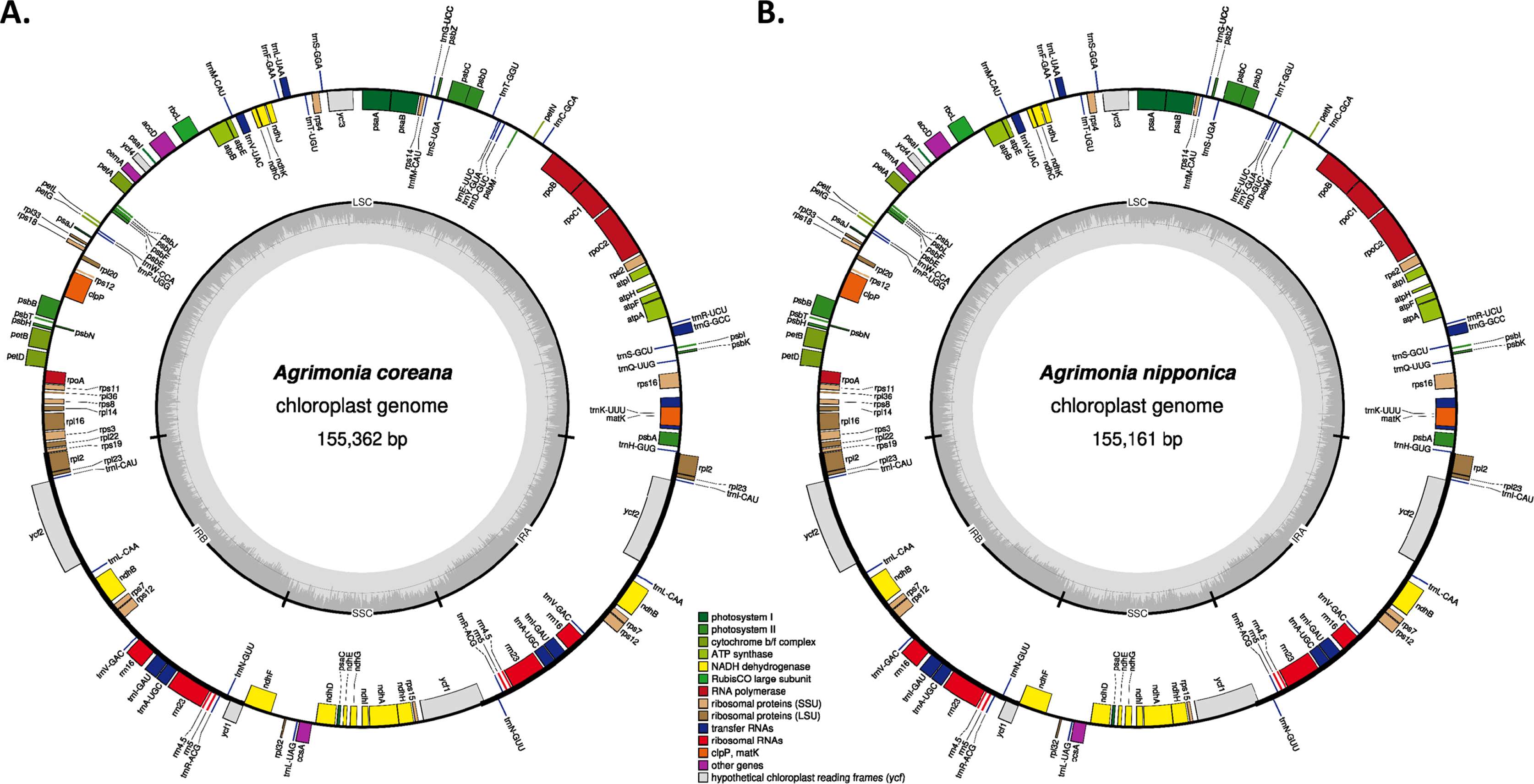
Circular gene map of Agrimonia coreana and Agrimonia nipponica chloroplast genomes. A. Agrimonia coreana, B. Agrimonia nipponica. Genes shown outside are transcribed clockwise, and those inside the circle are transcribed counter-clockwise. Genes are color-coded to distinguish different functional groups. The dark grey and the light grey plots in the inner circle correspond to the GC content and AT content, respectively.
The overall GC contents of A. coreana and A. nipponica amounted to 36.9%, identical to the remaining Agrimonia chloroplast genomes, except for the partial chloroplast genome (GenBank accession number: KY419942) (Table 1). The GC contents of the LSC in both A. coreana and A. nipponica amounted to 34.8%, whereas the corresponding SSC and IR rates of A. coreana were 30.3% and 42.6%, lower than those of A. nipponica (Table 1), caused by different range of the IR region.
Both of the Agrimonia chloroplast genomes contained 130 genes, consisting of 84 protein-coding genes (PCGs), eight ribosomal RNAs (rRNAs), 37 transfer RNAs (tRNAs), and one pseudogene (ycf1) (Online Supplementary Material S1).
The 17 genes duplicated in IR regions consisted of seven PCGs (rpl2, rpl23, ycf2, ndhB, rps7, rps12, and ycf1), four rRNAs (rrn16, rrn23, rrn4.5, and rrn5), and seven tRNAs (trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG, and trnN-GUU). The ycf1 gene in the IRb region was considered as a pseudogene because the putative start methionine was inside ndhF. However, ycf1 in A. pilosa var. nepalensis was annotated as a normal gene by the start methionine located at 89-bp inside ndhF, addressing the need for additional research on the correct position of ycf1 in the IR region. This caused one additional PCG in the A. pilosa var. nepalensis chloroplast genome (Table 1). In addition, the annotation of the three genes, rpl16, petD, and petB, in the A. pilosa var. nepalensis chloroplast indicated that they had no intron; however, those in the remaining Agrimonia chloroplast genomes consisted of two exons. The annotation of these genes should also be corrected for a more accurate analysis.
In the six Agrimonia chloroplast genomes, including the two Agrimonia chloroplast genomes sequenced in this study after correction of the corrected three genes in A. pilosa var. nepalensis, eight PCGs (rps16, rpoC1, petB, petD, rpl16, ndhB, ndhA, and rpl2) contain one intron and clpP, ycf3, and rps12 have two introns, which are conserved across Agrimonia chloroplast genomes. The gene structure of the six Agrimonia chloroplast genomes is well conserved.
Nucleotide diversity of six Agrimonia chloroplast genomes
To investigate the landscape of nucleotide diversity in six Agrimonia chloroplast genomes, nucleotide diversity from the alignment of the six Agrimonia chloroplast genomes was assessed (see Materials and Methods). The LSC and SSC regions displayed a high level of nucleotide diversity, whereas two IR regions exhibited a low level, congruent to those of other chloroplast genomes (Du et al., 2017; Jiang et al., 2017; Li et al., 2018, 2019; Liu et al., 2020a; Loeuille et al., 2021). Most highly diverse regions were intergenic regions, and some regions contained tRNA genes, specifically trnQ, trnS, trnT, trnS, and trnT (Fig. 2), which is a reasonable observation in that most genes in chloroplast genomes are essential for photosynthesis and self-replication (Online Supplementary Material S1). One exceptional case is ycf1, displaying high nucleotide diversity throughout the coding genes in comparison to other PCGs (Online Supplementary Material S2). In detail, 29 non-synonymous SNPs, 15 synonymous SNPs, and 9-bp insertions were found in ycf1 (Online Supplementary Material S2). The ratio of non-synonymous to synonymous SNPs (1.93) was high enough in comparison to those of all intraspecific variations of the PCGs of the C. album chloroplast genome (15:11 = 1.36), which is also considered as a high ratio in comparison to other plant species (Park et al., 2021d). In addition, ycf1 displays high nucleotide diversity within the genus (Hong et al., 2017; Liu et al., 2018; de Souza et al., 2019; Celiński et al., 2020; Loeuille et al., 2021) and even among certain species (Kim et al., 2020; Park et al., 2021d), which is also congruent with previous studies showing that ycf1 is the most promising plastid DNA barcode in land plants (Neubig et al., 2009; Dong et al., 2015). Interestingly, ycf1 is reportedly a target gene of PBR1 in Arabidopsis (Yang et al., 2016) as well as an essential part of the chloroplast protein import machinery (Kikuchi et al., 2013; de Vries et al., 2015). Moreover, ycf1 and ycf2 in the tobacco chloroplast genome reportedly have an essential function according to a transformation experiment (Drescher et al., 2000). Taken together, the selection pressure imposed on ycf1 may not be high enough in comparison to other PCGs in the chloroplast genome and appeared to be distributed evenly on the gene region (Online Supplementary Material S2), also addressing the need for further investigations of ycf1 through land plant species.
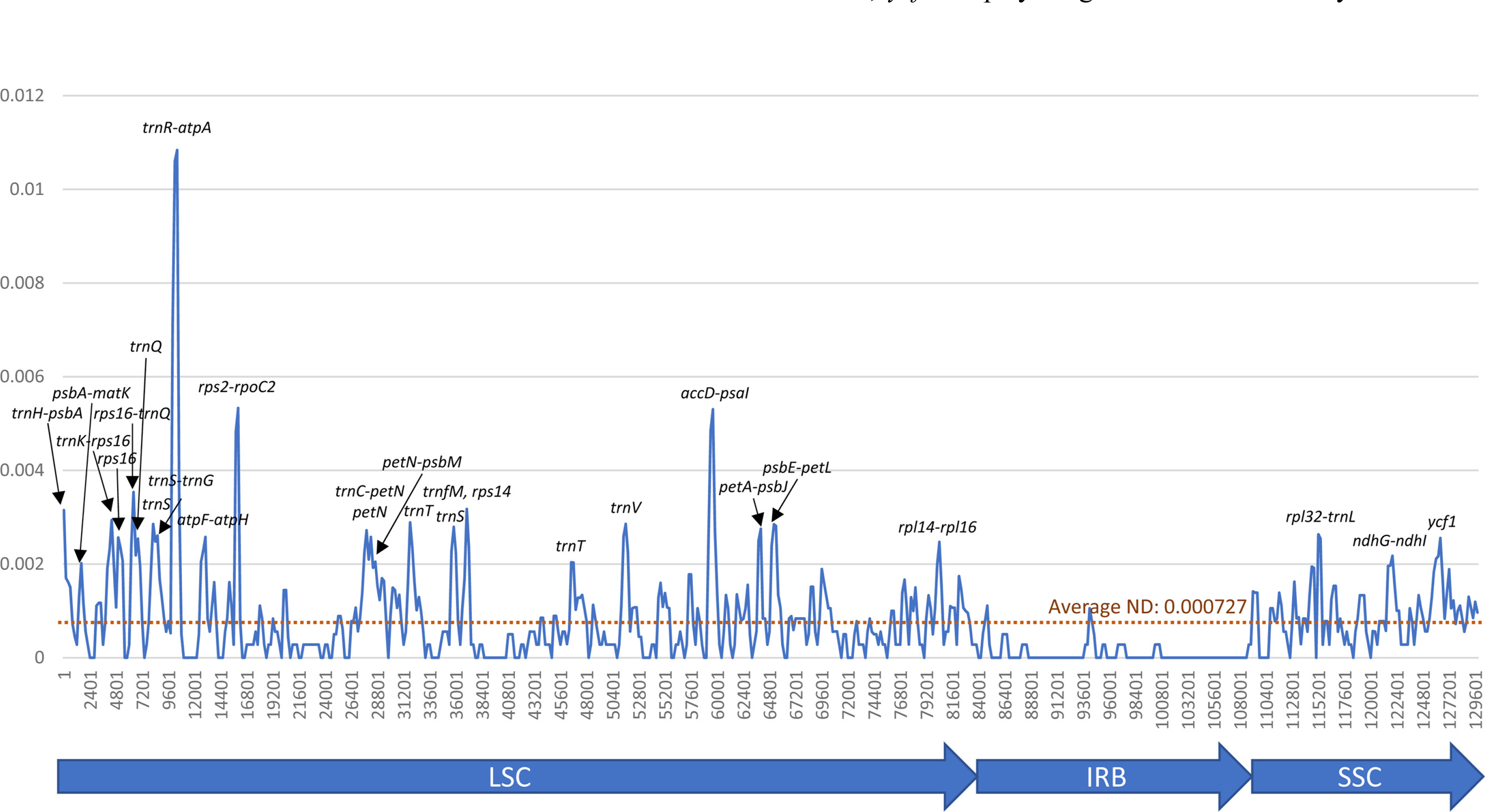
Nucleotide diversity of the six Agrimonia chloroplast genomes. The X-axis represents the coordination of the chloroplast genome and the Y-axis represents the nucleotide diversity values from 500-bp window sliding in 200-bp steps. Highest peaks of the nucleotide diversity graph display the names of the genetic regions. The three blue arrow bars below the X-axis indicate the large single-copy (LSC), inverted repeat (IR), and small single-copy (SSC) regions, respectively.
Intraspecific variations identified from the four complete chloroplast genomes of Agrimonia pilosa
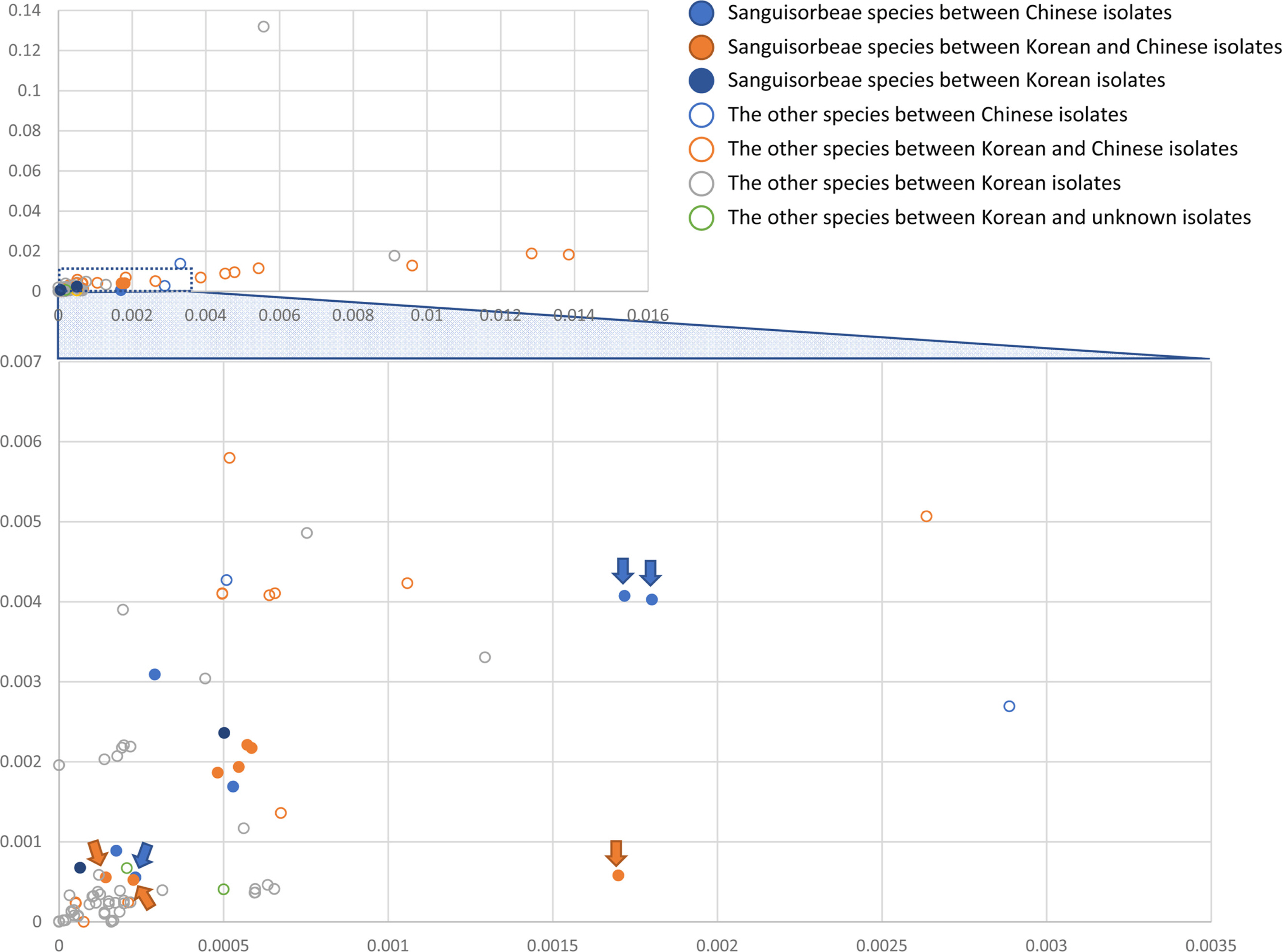
The intraspecific variations of the four A. pilosa complete chloroplast genomes were investigated based on pair-wise comparisons, as was done in previous studies (Kwon et al., 2019a; Park et al., 2019f, 2019h, 2021f; Oh et al., 2021), as this method offers advantage of estimating the degree of intraspecific variation. SNPs and INDELs identified from the four A. pilosa chloroplast genomes ranged from 22 to 280 and 16 to 105 (86 bp to 633 bp in length) (Fig. 3A). It should be noted that the partial chloroplast genome KY419942 (Zhang et al., 2017) exhibited a high degree of intraspecific variation against the remaining three chloroplast genomes, including different varieties of A. pilosa and A. pilosa var. nepalensis (Fig. 3B). This phenomenon was also found in two chloroplast genomes of Duchesnea indica (Andrews) Th. Wolf (Kim et al., 2020), among which one is also from the same study (Zhang et al., 2017). For a comparison of the degrees of intraspecific variation in other plant species calculated from various studies which investigated intraspecific variation in plant samples isolated in Korea and China (Table 2), the proportions of SNPs and INDELs against the length of the alignment sequences were calculated. This shows that the ratios of the SNPs range from 0.000226 to 0.001801 and those of the INDELs are from 0.000522 to 0.004073. The degrees of SNPs and INDELs for A. pilosa are separated by two groups (blue and orange arrows in Fig. 3B) due to the numerous variations found in KY419942 chloroplast genome (Fig. 3A).
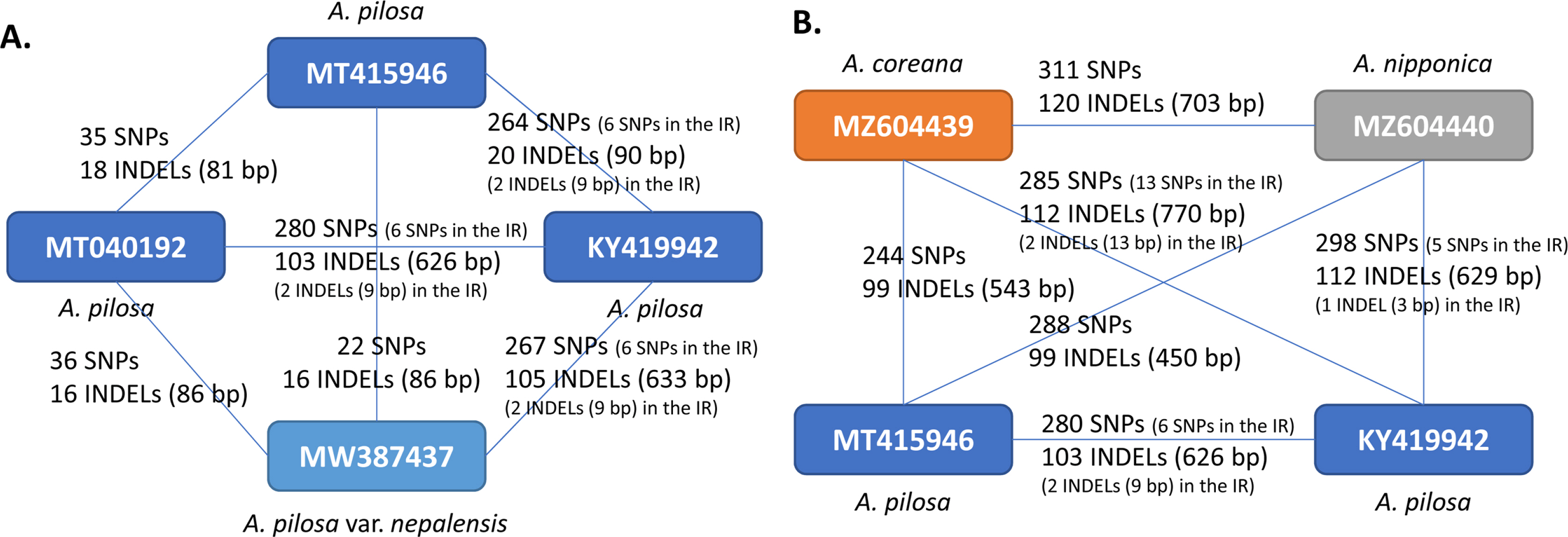
Plot of intraspecific variations identified from the chloroplast genome of various plant species: (A) display of the plot on which the Xaxis denotes the proportion of single nucleotide polymorphisms (SNPs) and the Y-axis is the proportion of insertions and deletions (INDELs) identified from various plant species. Three color-filled circles, shown in blue, orange, and dark blue, represent the Sanguisorbeae species, between Chinese isolates, between Korean and Chinese isolates, and between Korean isolates, respectively. Circles filled in with white represent the four groups, other species between Chinese isolates, those between Chinese isolates, those between Korean and Chinese isolates, those between Korean isolates, those between Korean and unknown isolates. B. Enlarged graph from the dotted lines in the graph in (A). Blue arrows and yellow arrows indicate the proportions of the SNPs and INDELs identified from A. pilosa chloroplast genomes.
To evaluate the degrees of SNPs and INDELs identified among available Agrimonia chloroplast genomes, the degrees of intraspecific variation from five species in the tribe Sanguisorbeae which contains more than one chloroplast genome were also identified (Table 3). Based on the geographical locations of the species in Sanguisorbeae, including Agrimonia species, three major groups were defined: (1) one within Chinese isolates, (2) one between Korean and Chines isolates, and (3) one within Korean isolates. The proportions of SNPs and INDELs identified from all three groups were distributed from low to high degrees (Fig. 3B), suggesting that these proportions are not strongly affected by the geographical distribution. This finding is also congruent with those associated with other plant species investigated in this study (Table 2); however, certain Orchidaceae species, including Goodyera schlechtendaliana Rchb. f. and Gastrodia electa Blume, displayed very high SNP and INDEL proportions (above 0.01) (Fig. 3A); these can be considered family-specific characteristics.
List of chloroplast genomes used for comparative analyses in this study.
Two hundred and ninety-two non-redundant SNPs identified based on a multiple-sequence alignment of four A. pilosa chloroplast genomes indicated that most of the SNPs are common among the SNPs identified based on six pair-wise comparisons of A. pilosa, as the largest number of SNPs among A. pilosa is 280, found between MT040192 and KY419942 (Fig. 3A). In addition, 78 out of 292 SNPs and two INDELs were found in 36 of 84 PCGs (42.85%), which is a large number in comparison with other intraspecific variations of Arabidopsis thaliana L. (11 PCGs) (Park et al., 2020f), C. album (nine PCGs) (Park et al., 2021d), and Campanula takesimana Nakai (eight PCGs) (Park et al., 2021a). It is remarkable that only three synonymous SNPs identified in three PCGs were from MT040192; two synonymous SNPs were from MT415946 and no variations were found in A. pilosa var. nepalensis (MW387437), indicating that the only five PCGs, matK, psbC, psaB, psbJ, and psbB, have only synonymous SNPs apart from KY419942, lower than those identified in the three species used in this comparison.
In the same sense, ycf1 exhibited 10 synonymous SNPs, 11 non-synonymous SNPs, and one 9-bp insertion, which did not cause a frameshift only from KY419942. It is also interesting that three A. pilosa chloroplast genomes but not KY419942 displayed no intraspecific variation in ycf1, which usually presents numerous intraspecific variations (Kim et al., 2020; Park et al., 2020f, 2021d). This provides indirect evidence of the distance between KY419942 and the remaining three A. pilosa chloroplast genomes.
An additional interesting factor is that certain proportions of SNPs and INDELs of Agrimonia species are highest among those of Sanguisorbeae species. Determination of these proportions was from pair-wise comparisons with the KY419942 chloroplast genome, which displays a high level of intraspecific variation (Fig. 3A). This difference supports the hypothesis that the KY419942 chloroplast genome evolved independently through, for instance, long-term geographical isolation, akin to island plant species (Zhang et al., 2019a; Celiński et al., 2020).
Interspecific variations identified from the chloroplast genomes of three Agrimonia species
Given the fewest intraspecific variations between A. pilosa and A. pilosa var. nepalensis among the six pair-wise comparisons of A. pilosa chloroplasts conducted here (Fig. 4A), the chloroplast genomes of A. coreana, A. nipponica, and A. pilosa were used to investigate the interspecific variations. Due to the differences from the KY419942 chloroplast genome (Figs. 4A), four Agrimonia chloroplast genomes were used for this investigation (Fig. 4B). The numbers of SNPs identified from the four chloroplast genomes range from 244 to 311 (Fig. 4B), higher than those of SNPs within A. pilosa (Fig. 4A) except for that between A. coreana and A. pilosa (GenBank accession number: MT415496). The numbers of INDEL regions and the total lengths of the INDEL regions identified from A. pilosa (GenBank accession number: MT415496) are slight lower than those identified from another A. pilosa chloroplast genome (GenBank accession number: KY419942) (Fig.4B). Moreover, ycf1, which has a high number of intraspecific variations in KY419942, presented more variations than the intraspecific variations of A. pilosa: 22 non-synonymous SNPs, 14 synonymous SNPs, and one 9-bp insertion. These variations show the presence of four non-synonymous SNPs and four synonymous SNPs from A. coreana; six non-synonymous SNPs and one synonymous SNP from A. nipponica; seven non-synonymous SNPs, five synonymous SNPs, and a 9-bp insertion from KY419942 (A. pilosa); and five non-synonymous SNPs and one synonymous SNP from MT415496 (A. pilosa). In addition, three synonymous SNPs showed that half were from A. coreana and MT415496 and half were from A. nipponica and KY419942, suggesting that KY419942 is a different species from A. pilosa based on the investigation of sequence variations, a case similar to certain cryptic plant species (Gurushidze et al., 2008; Okuyama and Kato 2009; Myszczyński et al., 2017; Li et al., 2020b; Liu et al., 2020b) or hybrid-origin species such as Arabidopsis suecica (r.) Norrl. (O'Kane et al., 1996; Novikova et al., 2017) and Arabis species (Kawabe et al., 2018).
Numbers of single nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs) identified among the four Agrimonia chloroplast genomes. Yellow, grey, and blue boxes indicate the A. coreana, A. nipponica, and A. pilosa chloroplast genomes, respectively. Numbers of SNPs and INDEL regions with the total lengths of INDEL regions are displayed on the lines connecting two chloroplast genomes. The comparisons with the KY419942 chloroplast genome exhibit additional information about the numbers of SNPs and INDELs in the IR region due to a lack of one of the IR regions in the KY419942 chloroplast genome.
The chromosome numbers for A. pilosa isolated in China and on Mt. Baekdu were reported to be 2n = 4x = 28 (Kwon et al., 2005) and 2n = 2x = 16 (Mitrenina et al., 2020), respectively. These different chromosomal configurations of A. pilosa suggest at least two possible origins of A. pilosa. First, a tetraploid of A. pilosa may have been formed via allopolyploidization, as in the case of Coffea arabica L. which originates from C. canephora Pierre ex A. Froehner and C. eugenioides S. Moore (Clarindo and Carvalho, 2008). In this case, two types of chloroplast genomes from two parental species can be found in a hybrid species if allopolyploidization occurred in both directions, in agreement with the two distinct types of chloroplast genomes found in this study. The second scenario involves two different specification events, resulting in two different basic chromosome numbers, n = 7 (Kwon et al., 2005) and n = 8 (Mitrenina et al., 2020). From the two speciation routes, two different types of chloroplast genomes could be passed down to A. pilosa, which can explain the current distinct between the two chloroplast genomes.
Phylogenetic analysis of the Agrimonia chloroplast genome sequence
ML and BI phylogenetic trees were constructed based on the 33 Sanguisorbeae species, including the two Agrimonia chloroplast genomes assembled in this study. Agrimonia exhibited monophyletic characteristics in that it formed one clade clustered with the Hagenia and Leucosidea genera (Fig. 5), congruent with the taxonomy of the subtribe Agrimoniinae (Chung, 2008) and previous phylogenetic analyses (Helfgott et al., 2000; Zhang et al., 2017). In addition, A. pilosa var. nepalensis was clustered with two A. pilosa complete chloroplast genomes isolated in Korea and China (Fig. 5), also in agreement with the low number of intraspecific variations among the four A. pilosa chloroplast genomes (Fig. 4A).
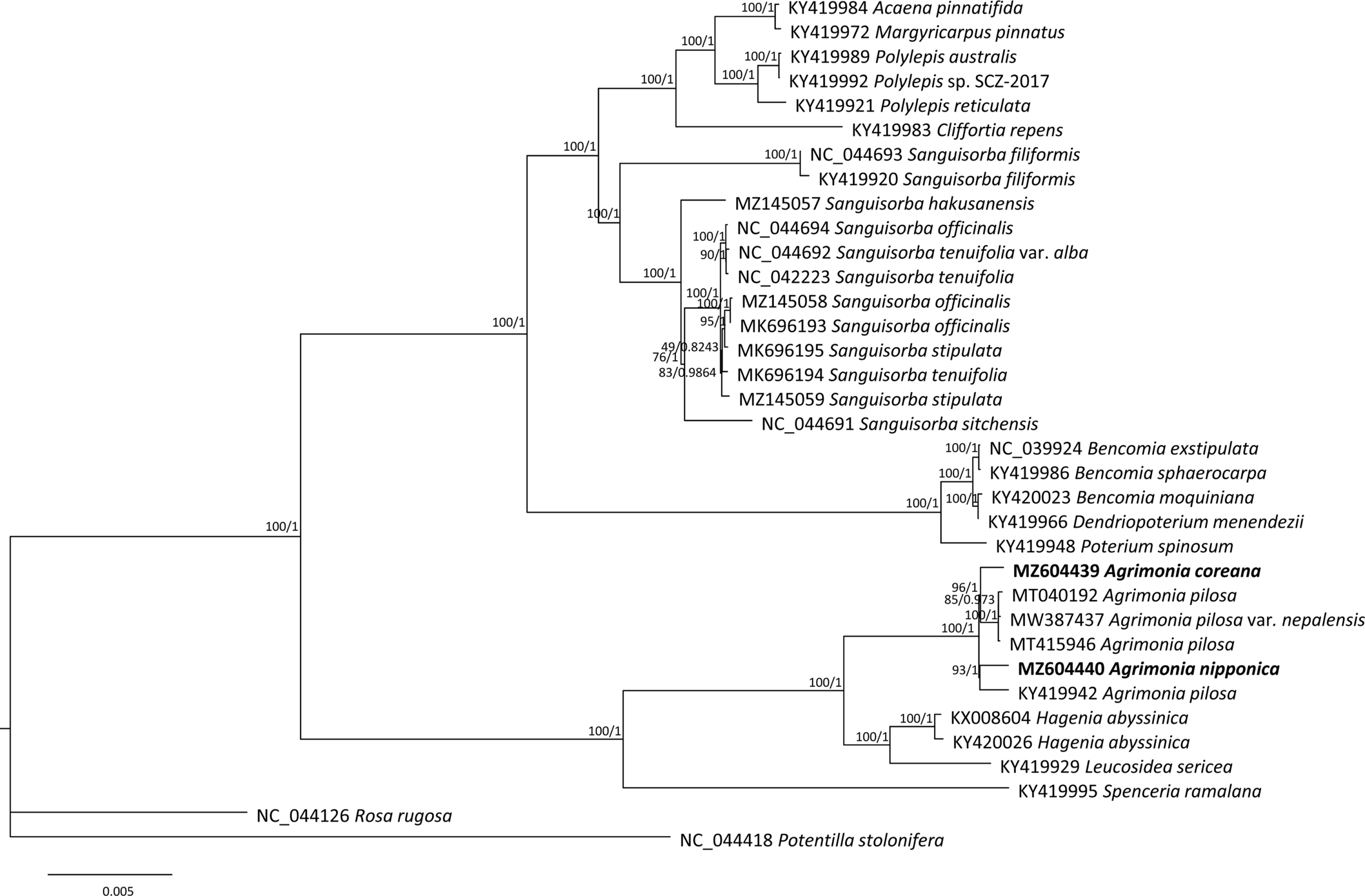
Maximum likelihood tree of 33 Sanguisorbeae chloroplast genomes with two outgroup species. Numbers on branches in the phylogenetic tree indicate the bootstrap values of ML and the prior possibility of Bayesian inference. Chloroplast genomes sequenced in this study are presented in bold text.
In contrast, the A. pilosa chloroplast (GenBank accession number: KY419942), which is different from the remaining three A. pilosa chloroplast genomes, is a sister to A. nipponica (Fig. 5), in contrast to the expected results based on the numbers of SNPs and INDELs (Fig. 3B). This indicates a need for an additional investigation of A. pilosa to test the suggested hypotheses that A. pilosa was formed from hybridization or independent speciation events. The chromosome numbers of the samples of A. pilosa from which chloroplast genomes were determined are unknown. The chromosome number heterogeneity of A. pilosa, as discussed above, associated with hybridization and polyploidization, may be responsible for the close relationship between the A. pilosa chloroplast (GenBank accession number: KY419942) and A. nipponica. The chromosome numbers of A. nipponica and A. coreana are 2n = 4x = 28 (Iwatsubo et al., 1993), reflecting the complex speciation history of Agrimonia species. Based on leaf morphological characteristics, A. coreana and A. nipponica are closely related by having 1–2 pairs of lateral leaflets in the mid-caule region and an abaxial surface of the leaflets with densely glandular and tomentose hairs (Chung, 2017), in contrast to the phylogenetic tree (Fig. 5). This indicates that additional research is needed to investigate the corresponding morphological features possibly to support the current phylogenetic tree or to consider the possibility of hybridized individuals of A. pilosa. Furthermore, A. pilosa may contain cryptic species, or there may even have been a misidentification of A. pilosa chloroplast genome (KY419942), as in the C. album (Park et al., 2021d) and Magnolia insignis (Wall.) Blume and Magnolia alba Figlar & Noot cases. Park (2020) can be referenced for further analyses.
Comparative analysis of SSR polymorphisms on six Agrimonia chloroplast genomes
SSRs were identified from the six Agrimonia chloroplast genomes, denoting the total numbers of SSRs, including normal SSRs, potential SSRs, and the extended SSRs ranging from 546 to 558 apart from KY419942 due to the lack of one IR region (Table 4). PentaSSR and hexaSSR identified in the chloroplast genome are usually classified into the potential SSR of which the length is 10 or 12 bp with two repeats. Only the A. coreana chloroplast genome contained two normal pentaSSRs (P0000056 and P0000057) with the three repeats (Fig. 6A). The numbers of normal SSRs for the six chloroplast genomes ranged from 73 to 83 (Fig. 6A). The four A. pilosa chloroplast genomes exhibited a range of 73 to 74, in good agreement with other plant species, such as C. album (53 to 55) (Park et al., 2021d). In addition, KY419942 without one IR region displayed the same level of normal SSRs (Fig. 6A), reflecting that the IR region of A. pilosa does not contain normal SSRs. The A. coreana and A. nipponica chloroplast genomes contained more normal SSRs than those of A. pilosa: A. coreana covers more monoSSRs and pentaSSRs, while A. nipponica had more diSSRs and tetraSSRs (Fig. 6A).
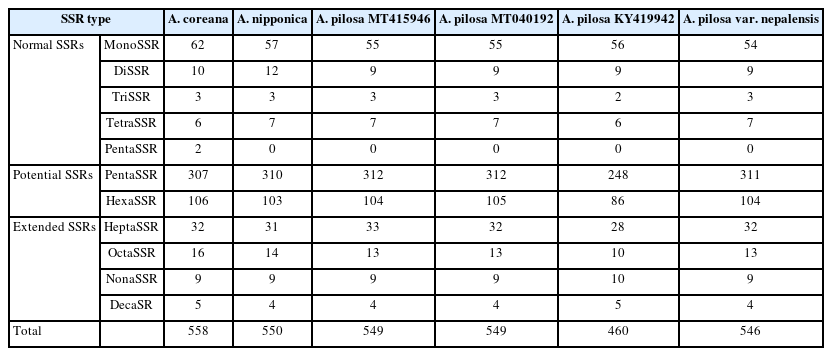
List of normal SSRs, potential SSRs, and extended SSRs identified from the six Agrimonia chloroplast genomes.
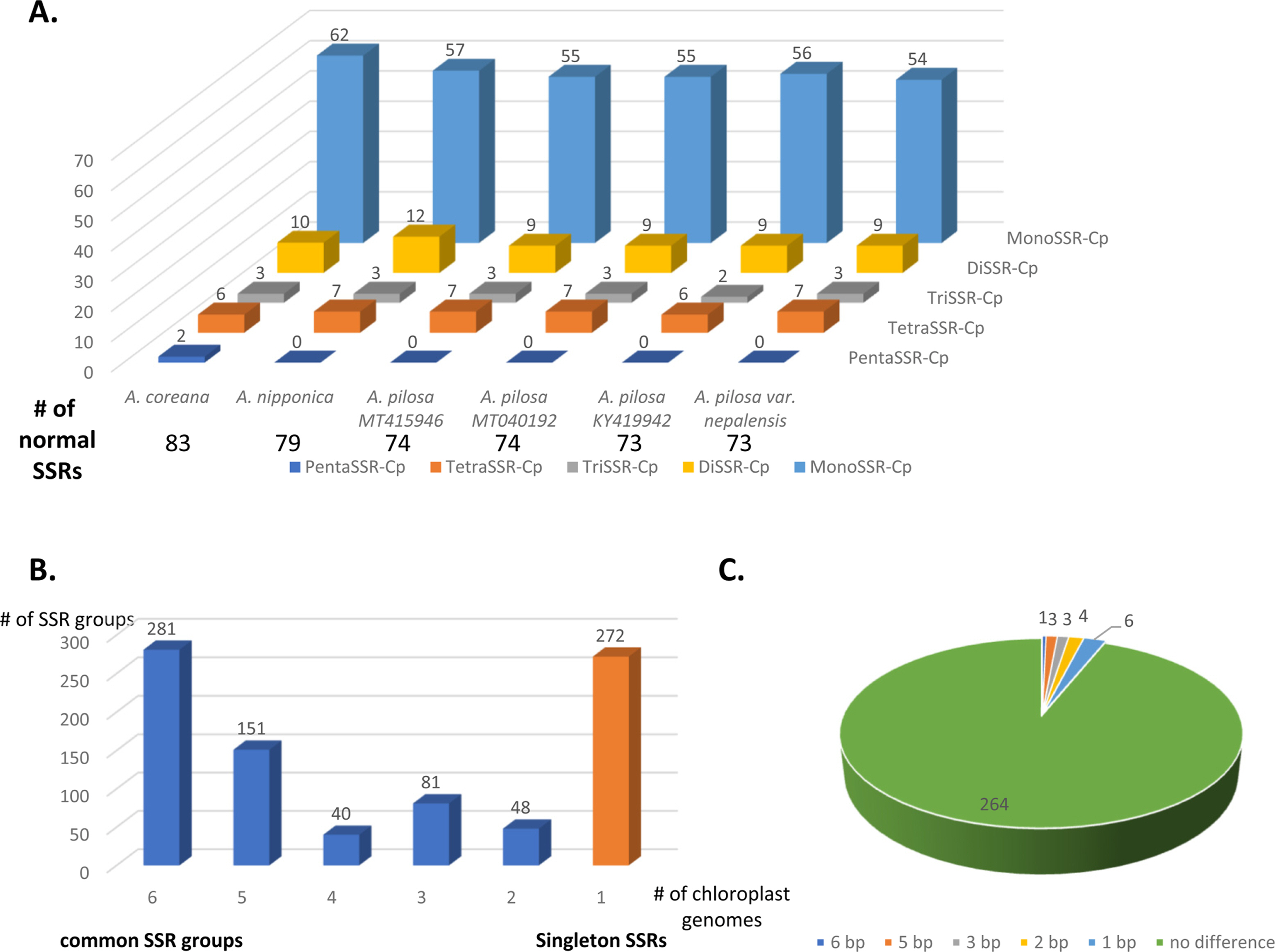
Number of normal simple sequence repeats (SSRs) and distribution of SSR groups identified on six Agrimonia chloroplast genomes. A. Number of SSR types in each of the six Agrimonia chloroplast genomes. The X-axis represents the six Agrimonia chloroplasts and the Yaxis indicates the number of normal SSRs. The five different colors indicate the five types of SSRs, i.e., monoSSRs, diSSRs, triSSRs, tetraSSRs, and pentaSSRs. The table below the X-axis represents the numbers of normal SSRs along with the six Agrimonia samples. B. Number of SSR groups along with the number of samples in each SSR group. The X-axis is the number of species in the SSR groups and the Y-axis is the number of SSR groups. The orange-colored bar indicates singleton SSRs. C. Pie graph presenting SSR groups, showing the length differences in the SSR group.
Based on a comparison of the flanking sequences of SSRs identified from the six Agrimonia chloroplast genomes, 601 SSR groups and 272 singleton SSRs were identified (Fig. 6B). In addition, 281 of 601 SSR groups (46.76%), covering six SSRs from six different chloroplast genomes, were identified (Fig. 6B). This indicates that fewer than half of the Agrimonia SSRs are shared among the six chloroplast genomes, referred to as common SSR groups. One hundred and fifty-one SSR groups (25.12%) contained five SSRs that originated from five chloroplast genomes (Fig. 6B). Seventeen out of 281 common SSR groups (6.05%) exhibited length variations in the SSR regions (Fig. 6C), representing a good molecular marker candidate with which to distinguish species or populations. The length differences in the SSR region ranged from 1 bp to 6 bp (Fig. 6C). Twelve out of 17 common SSR groups (70.59%) were monoSSR and the remaining five common SSR groups consisted of two diSSRs, two pentaSSRs, and one pentaSSR/hexaSSR (Table 5). The greatest difference (6 bp found in SSR Group 281) was caused by three additional time repeats of diSSRs, indicating that the SSRs in KY419942 and the A. coreana chloroplast genomes were eight-fold TA diSSR while the remaining SSRs are five-fold TA diSSR (Table 5). Interestingly, SSR Group 189 displayed one of six SSRs that was the pentaSSR type (A. nipponica), while the remaining SSRs were hexaSSRs (Fig. 6B, C). These variations in the 17 common SSR groups can serve as a good example for developing molecular markers based on SSRs in chloroplast genomes. In detail, the 151 SSR groups covering five Agrimonia chloroplast genomes can be considered as another type of molecular marker candidate because one of the six chloroplast genomes lacks the SSR, which is clear evidence allowing the detection of a specific Agrimonia sample. Moreover, because the number of SSRs can be affected by the evolution of chloroplast genomes (Sawicki et al., 2020), SSRs identified from the six Agrimonia chloroplast genomes can be used to understand the corresponding evolutionary features with the additional chloroplast genome of Agrimonia and a neighbor genus in the near future. Taken together, the variations identified in the SSRs from six Agrimonia chloroplast genomes exhibit not only the dynamic characteristics of Agrimonia SSRs but also suggest potential uses of Agrimonia chloroplast genomes.
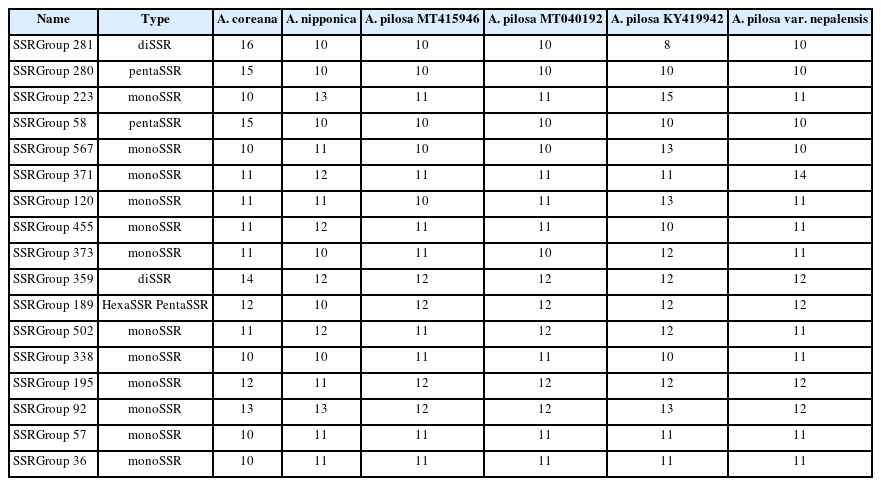
List of SSRs from common SSR groups displaying length difference of SSR regions.
CONCLUSION
We completed the chloroplast genomes of A. coreana and A. nipponica isolated in Korea and conducted comparative analyses of six Agrimonia chloroplast genomes originating from three species. The Agrimonia coreana and A. nipponica chloroplast genomes are 151,362 bp and 155,161 bp long, respectively. High nucleotide diversity was detected in the ycf1 gene, congruent with earlier work (Hong et al., 2017; Jiang et al., 2017; Liu et al., 2018; de Souza et al., 2019; Kim et al., 2019d; Li et al., 2019; Celiński et al., 2020; Park et al., 2020e; Loeuille et al., 2021). The intraspecific variations of four A. pilosa chloroplast genomes revealed that KY419942 is distinct from the remaining three A. pilosa chloroplast genomes. In addition, the interspecific variations among A. coreana, A. nipponica, and A. pilosa indicated that the distance between two A. pilosa chloroplast genomes is similar to that between A. coreana and A. nipponica. A phylogenetic analysis found that KY419942 was clustered with A. nipponica, suggesting that KY419942 (A. pilosa) might have been misidentified or a cryptic species caused by complex evolutionary histories. A comparison of SSRs identified from the six Agrimonia chloroplast genomes suggests potential molecular markers with which to distinguish between species or among populations of the same species. Considering these aspects together with the results of this study, our A. coreana and A. nipponica chloroplast genomes provide insight into the phylogenetic relationships among Agrimonia species in Korea and about the morphological as well as interspecific and intraspecific features of chloroplast genomes, including variations of chloroplast genomes. Moreover, information about the two Agrimonia chloroplast genomes serves to unravel the complex evolutionary history of A. pilosa, A. coreana, and A. nipponica, which require further analyses that can include deciphering additional chloroplast genomes of A. pilosa and neighboring species.
ONLINE SUPPLEMENTARY MATERIALS
S1 and S2 are available at https://doi.org/10.11110/kjpt.2024.54.1.147.
Nucleotide diversity of ycf1 in the six Agrinonia chloroplast genomes
X-axis presents coordination of chloroplast genome and Y-axis presents values of nucleotide diversity value of 500-bp window sliding by 200-bp step displaying ycf1 genic regions. Below X-axis, positions of sequence variations identified in ycf1 were presented as black lines in the six grey bars, corresponding to the six Agrinonia chloroplast genomes. Blue-colored circles under the gene diagram means synonymous SNPs and orange-colored circles indicate non-synonymous SNPs. INDEL was depicted as blue-colored triangle.
Numbers and proportions of SNPs and INDELs identified among the four Agrimonia pilosa chloroplast genomes
(A) Blue boxes indicate A. pilosa chloroplast genomes and light-blue box means A. pilosa var. nepalensis. Number of SNPs and INDEL regions with total length of INDEL regions were displayed on the lines connecting two chloroplast genomes. The comparisons with KY419942 chloroplast genome exhibit additional information of # of SNPs and INDELs in the IR region due to lack of one of IR regions in KY419942 chloropalst genome. (B) Blue boxes indicate A. pilosa chloroplast genomes and light-blue box means A. pilosa var. nepalensis. Proportion of SNPs and INDEL regions against the alignment length and alignment length were displayed on the lines connecting two chloroplast genomes.
Acknowledgements
We would like to express our appreciation of Dr. Sang-Hun Oh for the useful advice about this manuscript. This work was supported by an InfoBoss Research Grant (IBG-0035).
Notes
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.