Viburnum L. (Adoxaceae) widely distributed in temperate and subtropical regions of the Northern Hemisphere encompassing Europe, North Africa, Asia, and the Americas includes 175–200 species of small trees and shrubs (Rehder, 1908; Donoghue, 1983; Hara, 1983; Donoghue et al., 2004). There are ten species of the genus in Korea (Choi et al., 2018; Choi and Oh, 2019). The V. dilatatum complex in Korea consists of four morphologically similar species, i.e., V. dilatatum Thunb., V. erosum Thunb., V. japonicum (Thunb.) C. K. Spreng., and V. wrightii Miq. It is characterized by free bud scales and serrate leaves with pinnate veins and extrafloral nectaries at base on the abaxial surface (Choi and Oh, 2019). Phylogenetic analysis of Viburnum based on the nuclear internal transcribed spacers (ITS) regions and chloroplast rbcL, matK, and psbA-trnH regions strongly supported the complex as monophyletic but the relationships among the species within the complex remain unclear (Choi et al., 2018). Viburnum japonicum was supported as a monophyletic group in the ITS data but unresolved with V. dilatatum, V. erosum, and some accessions of V. wrightii. The cpDNA data showed one or two substitutions among the individuals of the complex included in the analysis.
Recent applications of next-generation sequencing (NGS) tools have generated the rapid accumulation of genomic data (Metzker, 2010; Bleidorn, 2016; Goodwin et al., 2016). The completion of chloroplast genomes along with comparative analysis methods have become easier than ever before as the number of reference genomes increases. A complete chloroplast genome can serve as a tool during the development of markers for phylogenetic (Nikiforova et al., 2013; Clement et al., 2014; Zimmer and Wen, 2015; Hou et al., 2016) and phylogeographic analyses (Lexer et al., 2013; Zimmer and Wen, 2015). However, not all NGS data have been assembled into complete chloroplast genomes due to technical difficulties or the presence of complex structures, such as repetitive sequences. Genes and non-coding regions from taxa studied are extracted to the greatest extent possible and are used in phylogenetic analyses.
Complete chloroplast genome sequences were reported from two species in the V. dilatatum complex: V. japonicum (Cho et al., 2018) and two different accessions of V. erosum (Park et al., 2019a; Choi et al., 2020). Chloroplast genomes can be reconstructed when there are sufficient numbers of raw reads deposited in public databases, such as those managed by the National Center for Biotechnology Information (NCBI), European Bioinformatics Institute (EBI), or DNA Data Bank of Japan (DDBJ). This applies to the process in which some of the chloroplast gene sequences of twenty-two Viburnum species were stored in Short Reads Archive (SRA) in the NCBI database (PRJNA239994). Seventy-three coding regions (52,758 bp in total) and 51 non-coding regions (16,819 bp in total) were recovered from 22 species and were used in a phylogenetic analysis of Viburnum (Clement et al., 2014). Their focus was on phylogenetic relationships among the species of Viburnum based on sequences from the chloroplast genome, as opposed to assembling the chloroplast genome itself. These raw reads are important resources for those attempting to assemble the complete sequence of the chloroplast genome, generating new information about structural variations, additional nucleotide sequences for phylogenetic analyses, and comparative genomics for a better understanding of the evolution of Viburnum. In particular, SRA contain raw reads from V. dilatatum which could be used in comparative genomics of the V. dilatatum complex. Recent phylogenomic analysis of Dipsacales (Xiang et al., 2019) and Caprifoliaceae s.l. (Wang et al., 2020) included Viburnum, but species of the V. dilatatum complex were not represented.
This study aims to establish a procedure to use when assembling a complete chloroplast genome from raw reads deposited in the NCBI database, to compare the chloroplast genomes of the V. dilatatum complex along with other species in Viburnum examining the level of variations among the species, and to reconstruct phylogenetic relationships of the species in the complex inferred from the entire genome of the chloroplast.
Materials and Methods
Chloroplast genome assembly and annotation
As the first step in this effort, the raw sequences derived from V. dilatatum and V. amplificatum Kern, which is used a distantly related lineage relative to the V. dilatatum complex, were used to reconstruct the whole chloroplast genome (Clement et al., 2014).
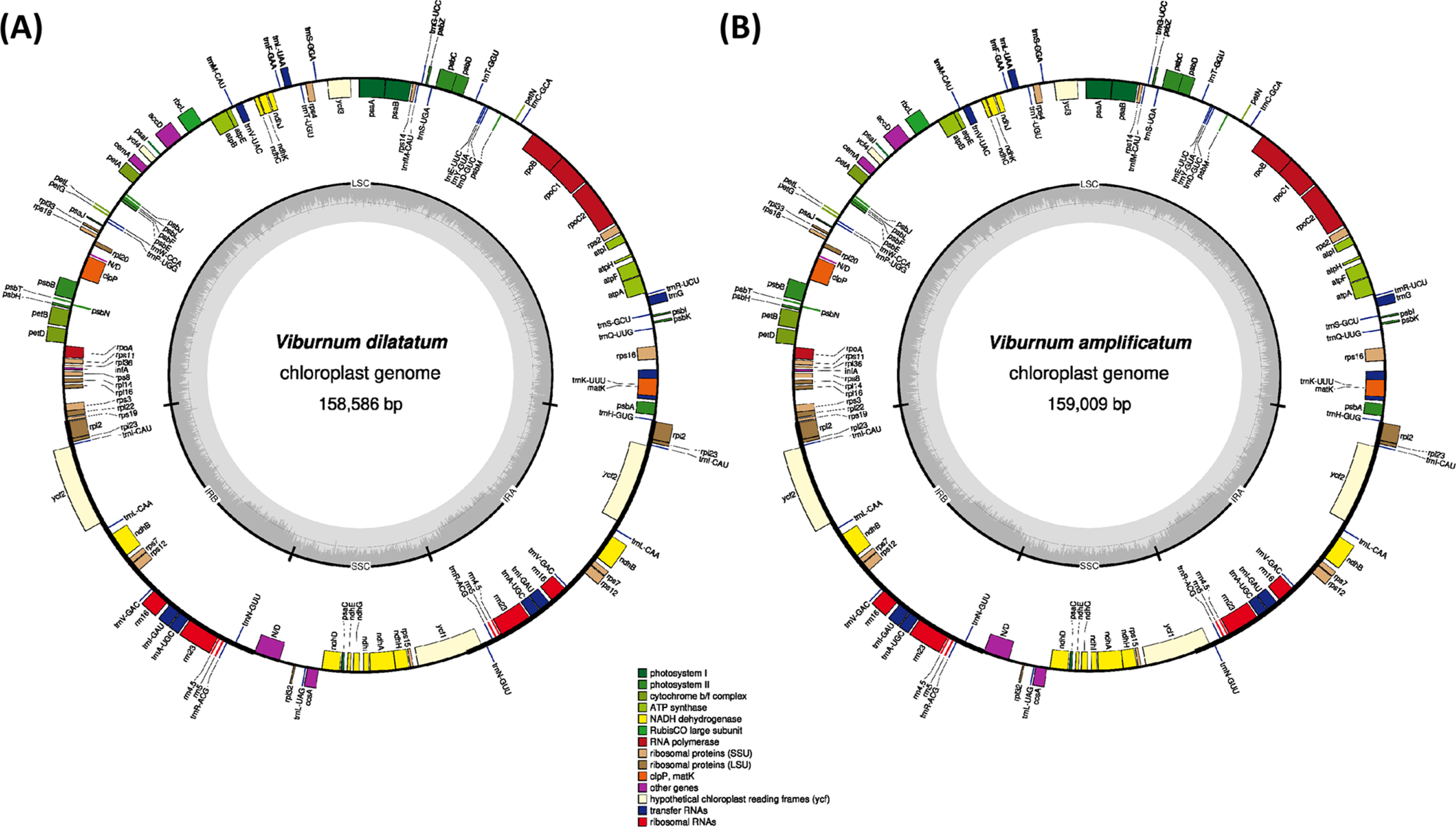
Raw sequences were downloaded from the SRA in the NCBI database (accession no. SRR1282417 for V. dilatatum and SRR1282983 for V. amplificatum) and filtered using Trimmomatic 0.33 (Bolger et al., 2014). The filtered paired-end reads were de novo assembled using Velvet 1.2.10 (Zerbino and Birney, 2008) with multiple k-mers ranging from 51 to 81 to select the best assembly result. Gap filling was conducted with GapCloser of SOAPdenovo 1.12 (Zhao et al., 2011). Assembled sequences were confirmed using BWA 0.7.17 (Li, 2013) and SAMtools 1.9 (Li et al., 2009) to correct misassembled bases. The tRNAs were confirmed using tRNAscan-SE (Lowe and Eddy, 1997). Annotation was conducted using Geneious R11 11.0.5 (Biomatters Ltd., Auckland, New Zealand) with the Viburnum erosum chloroplast genome (GenBank accession no. MN218778) (Park et al., 2019a), and the annotated chloroplast genome sequences were submitted to GenBank. The annotated GenBank format sequence file was used to draw a circular map using OGDRAW 1.3.1 (Greiner et al., 2019) with the default options.
Nucleotide diversity of Viburnum chloroplast genomes
The complete chloroplast genomes of V. dilatatum and V. amplificatum were compared to those of the other available Viburnum species (five chloroplast genomes from four species) by aligning these sequences with MAFFT 7.450 (Katoh and Standley, 2013). Based on these alignments, nucleotide diversity was calculated using the method proposed by Nei and Li (Nei and Li, 1979) implemented in the Plant Chloroplast Database (PCD; http://www.cp-genome.net) (Park et al., in preparation). The window size was set to 500 bp and the step size to 200 bp in the sliding-window analysis. The genomic coordination of each window was compared with gene annotation of the chloroplast genome to understand the features of the nucleotide diversity indexes.
Phylogenetic analysis
Complete chloroplast genomes of eight chloroplast genomes from six Viburnum species and Sambucus williamsii, used as an outgroup, were aligned using MAFFT 7.450 (Katoh and Standley, 2013). All chloroplast genome sequences were retrieved from the PCD (http://www.cp-genome.net) (Park et al., in preparation). The maximum-likelihood (ML) trees were reconstructed in MEGA X (Kumar et al., 2018). During the ML analysis, a heuristic search was used with nearest-neighbor interchange (NNI) branch swapping, the Tamura-Nei model, and uniform rates among sites. All other options were set to their default values. Bootstrap analyses with 1,000 pseudoreplicates were conducted with the same options. The posterior probability of each node was estimated by means of the Bayesian inference using MrBayes 3.2.7a (Huelsenbeck and Ronquist, 2001). The General Time Reversible model with gamma rates was used as a molecular model. A Markov-chain Monte Carlo algorithm was employed for 1,000,000 generations, sampling trees every 200 generations, with four chains running simultaneously. Trees from the first 100,000 generations were discarded as burn-in.
Results and Discussion
Complete chloroplast genome of V. dilatatum and V. amplificatum
The complete chloroplast genomes of V. dilatatum and V. amplificatum were successfully assembled from the raw sequences deposited in the Sequence Read Archive of the NCBI database (SRR1282417 and SRR1282983; GenBank accessions are MT165523 and MN720218, respectively). Number of raw reads of V. dilatatum and V. amplificatum was 13,064,398 and 23,639,938, respectively. These raw data provided 432-fold coverage for the chloroplast genome of V. dilatatum and 161-fold for V. amplificatum, which were sufficient amount to complete the whole genome. The chloroplast genome of V. dilatatum is 158,586 bp in length and has four subregions: 87,064 bp of large single copy (LSC) and 18,492 bp of small single copy (SSC) regions, along with 26,515 bp of a pair of inverted repeats (IRs) (Table 1). The GC ratio of the complete chloroplast genome is 38.1% and those of the LSC, SSC, and IR regions are 36.4%, 32.0%, and 43.0%, respectively. The length of chloroplast genome of V. amplificatum is 159,009 bp with the four subregions: 87,545 bp of LSC and 18,518 bp of SSC regions, along with 26,473 bp of a pair of IRs (Table 1). The GC ratio of the complete chloroplast genome is 38.1% and those of the LSC, SSC, and IR regions are 36.3%, 32.1%, and 43.0%, respectively.
The structure and gene content of the two newly assembled chloroplast are identical to those of V. erosum and V. betulifolium with 130 genes consisting of 85 protein-coding genes (PCGs), 37 transfer RNAs, and eight ribosomal RNAs (Table 2). Seventeen genes consisting of seven tRNA genes (trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-AGC, and trnN-GUU), four rRNA genes (rRNA5, rRNA4.5, rRNA23, and rRNA16), and six PCGs (ndhB, rps12, rpl23, ycf2, rps7, and rpl2) are duplicated in the IR regions. Eleven genes (trnK-UUU, rps16, atpF, rpoC1, petB, petD, rpl2, ndhB, trnI-GAU, trnA-UGC, and ndhA) contain one intron, and clpP, rps12, and ycf3 have two introns. In addition, the complete ycf1 gene in both chloroplast genomes are located in the IR region at the SSC/IR junction (Fig. 1).
The numbers of PCGs and tRNAs of V. dilatatum and V. amplificatum differ slightly from those of V. japonicum and V. utile (Table 2). Specifically, V. japonicum has the ndhF gene, which is not found in the other six chloroplast genomes. In addition, the chloroplast of V. japonicum does not contain rpl22. The Viburnum utile chloroplast does not have the ndhH, rps3, and rpl22 PCGs; however, it contains orf42 and orf188, which are not found in the remaining five chloroplast genomes. Viburnum erosum, V. japonicum, V. dilatatum, and V. betulifolium are members of the Succodontotinus clade, which is more closely related to V. amplificatum in the Lutescentia clade than it is to V. utile belonging to the Lantana clade. Thus, the addition of ndhF and the loss of rpl22 in V. japonicum should have occurred in the lineage of the species. The absence of rpl22 in V. japonicum and V. utile suggests that the loss of rpl22 should have occurred independently within Viburnum. These results imply that the conservation of gene contents may be relaxed in Viburnum. However, more complete taxon sampling is required to understand the evolution of the chloroplast structure in Viburnum.
The chloroplast genome of V. amplificatum shows the longest chloroplast genome, as it has the longest LSC among the seven (Table 1); while V. dilatatum displays the third shortest chloroplast genome among the seven available Viburnum chloroplast genomes (Table 1). In contrast, the chloroplast genome of V. utile is the shortest, and it has the shortest LSC (Table 1). The SSC of V. amplificatum is slightly longer than one of two accessions of V. erosum (GenBank accession no. MN641480), which is the second shortest among the seven genomes (Table 1). The length difference among the species of the V. dilatatum complex ranged from 1 bp between V. dilatatum and V. erosum (MN641480) to 38 bp between V. dilatatum and V. erosum (MN218778). The length differences between the species of the V. dilatatum complex and other species in Viburnum were much high, ranging from 385 (V. erosum vs. V. amplificatum) to 1,004 bp (V. erosum vs. V. utile). Thus it can be concluded that chloroplast genomes of the species in the V. dilatatum complex are similar to each other in length.
Interspecific sequence variations in the V. dilatatum complex
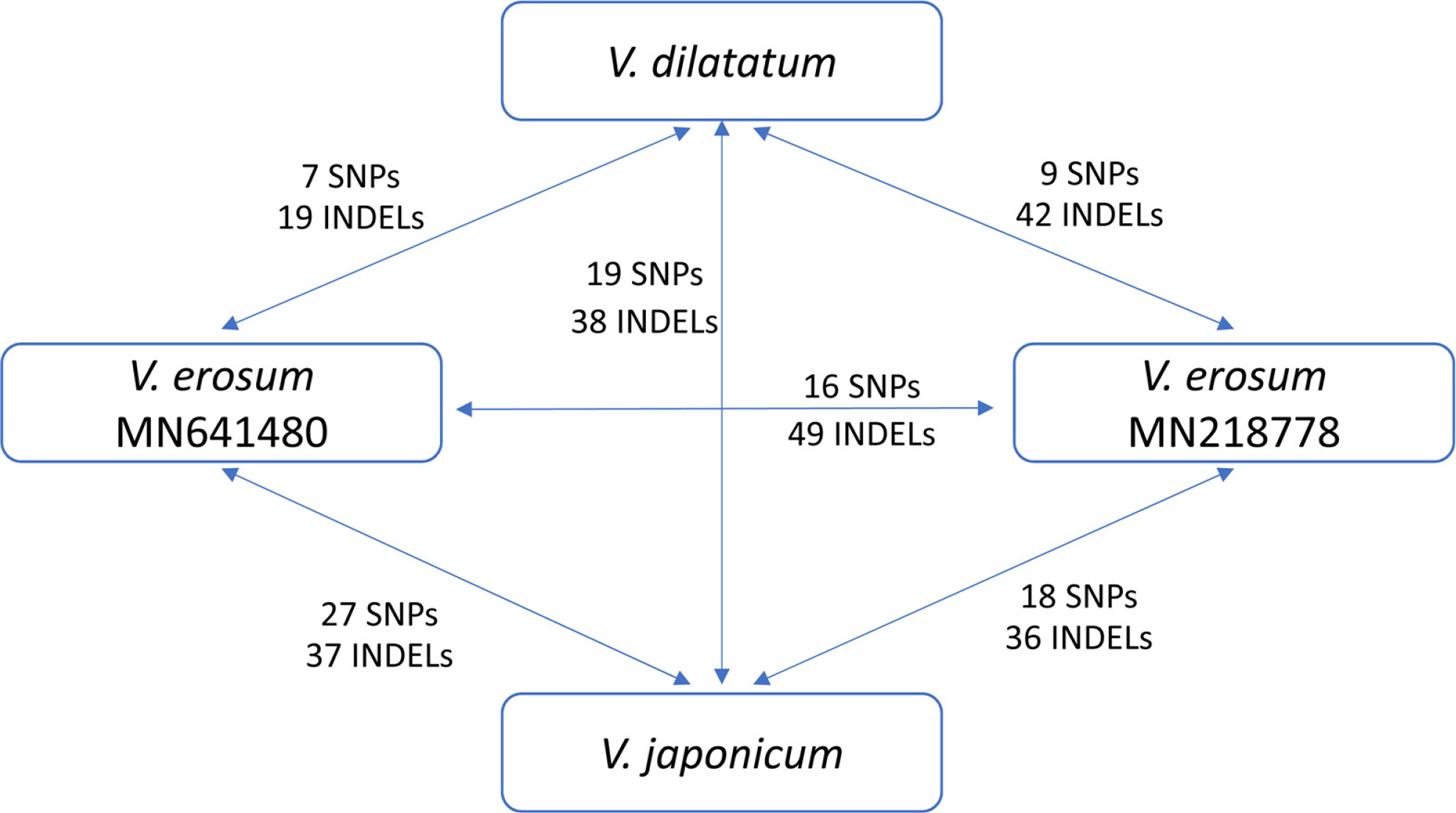
The level of the interspecific variations of the chloroplast genome of the V. dilatatum complex in terms of the number of single nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs) was low (Fig. 2). In this case, 7 SNPs and 19 INDELs and 9 SNPs and 42 INDELs were identified between V. dilatatum and each of the two accessions of V. erosum, respectively while 19 SNPs and 38 INDELs were found between V. dilatatum and V. japonicum. The numbers of SNP and INDEL within V. erosum are 16 and 49, suggesting that the intraspecific variation of chloroplast genomes of V. erosum is higher than interspecific variation between V. erosum and V. dilatatum (Fig. 2).
The overall number of interspecific variations in the V. dilatatum complex are much lower level than those of comparison of other species in Viburnum. For example, 1,021 SNPs and 1,697 INDELs were found between V. dilatatum and V. amplificatum. Similarly, 944 SNPs and 3,080 INDELs were identified between V. amplificatum and V. utile, and 1,295 SNPs and 2,933 INDELs were found between V. amplificatum and V. betulifolium. Thus, the low level of interspecific variation within the V. dilatatum complex suggests that the species in the complex may have been differentiated recently.
Nucleotide diversity among six Viburnum species
The nucleotide diversity among seven Viburnum chloroplast genomes was 0.00176 on average (Fig. 3A), a value ca. four times lower than that of the three Dysphania chloroplast genomes (0.0068) (Kim et al., 2019b). Species of Viburnum are woody plants that have longer generation times than annual or perennial plants, such as Dysphania. The low level of nucleotide diversity in Viburnum can be explained by its life history, specifically by its longer generation times that promote the conservation of the chloroplast genome (Smith and Donoghue, 2008). However, five out of seven plastomes in the comparison are from closely related species in the Succodontotinus clade, including the V. dilatatum complex. Sampling may have resulted in seemingly biased nucleotide diversity. Based on four regions of chloroplast genomes (LSC, SSC, and two IRs), the two IR regions present the lowest nucleotide diversity, indicating that the IR regions are highly conservative. Except for the region (from 29,800 bp to 31,200 bp), which displays very high nucleotide diversity (Fig. 3A), SSC presents the highest nucleotide diversity, followed by LSC.
Unlike other studies of the nucleotide diversity of chloroplast genomes such as Dysphania (<0.03) (Kim et al., 2019b) and Chenopodium (with one highly variable region showing the highest peak in an IR region with pi = 0.07885) (Hong et al., 2017), Rosa (<0.02) (Jeon and Kim, 2019), and Symplocarpus (<0.03) (Kim et al., 2019a), there is a group of high peaks on the Viburnum chloroplast genomes (29,800 to 31,200 bp; denoted by the blue circle labeled as 1 in Fig. 3A). This highly variable region located in LSC contains two large gap regions (Fig. 3B). Gap region 1 was found in V. dilatatum, V. erosum, and V. japonicum, and it represented large deletions in the species. Presence of the gap region 1 in the species of V. dilatatum complex but not in V. betulifolium, all of which are members of the Succodontotinus clade, suggesting that the species of the V. dilatatum complex are more closely related to each other than they are to V. betulifolium. A previous phylogenetic study did not resolve the relationship (Clement et al., 2014), thus the presence of gap region 1 would be a potential synapomorphy for the V. dilatatum complex.
The presence of gap region 2 in V. utile and V. betulifolium, which are members of the Lantana and Succodontotinus clade, respectively, suggests that deletions in gap region 2 may have occurred independently. Alternatively, the insertion of nucleotides in gap region 2 may have occurred in V. amplificatum and in an ancestor of the V. dilatatum complex independently. These long gaps on the chloroplast genome are also found in Coffea arabica (Park et al., 2019e), Duchesnea chrysantha (Park et al., 2019c), and Illicium anisatum (Park et al., 2019d). This type of gap was also identified on the mitochondrial genomes of Liriodendron tulipifera (Park et al., 2019b) and Populus alba × glandulosa (Park et al., 2019f).
In addition, second and third peaks representing highly variable regions are found in the boundary between IR and SSC regions (blue circles labeled as 2 and 3 in Fig. 3A); the second-highest peak is just after the end of the IR region, in an inter-exonic region. The third-highest peak arises at the end of the IR boundary, which is the end of the rrn23 gene, similar to the highest peak of Chenopodium study (Hong et al., 2017).
Phylogenetic analyses of chloroplast genomes of the V. dilatatum complex
The data matrix of the entire chloroplast genomes from seven accessions of Viburnum and Sambucus williamsii, an outgroup, included 163,153 sites, of which 5,221 were variable and 753 were parsimoniously informative. Phylogenetic analyses of chloroplast genomes of the V. dilatatum complex using ML and Bayesian methods showed that V. dilatatum is sister to one of two accessions of V. erosum, making V. erosum paraphyletic (Fig. 4). The two accessions of V. erosum were from Korea (Choi et al., 2020; Park et al., 2019a), while the original source of V. dilatatum was unknown, derived from cultivated plant on the Yale University campus (Clement et al., 2014). It is unclear whether the close relationship of V. dilatatum and V. erosum (GenBank accession no. MN641480) may have derived from a geographic factor. Previous analysis of three chloroplast regions (Choi et al., 2018) did not show any resolution among the species of the complex. The phylogenetic analysis of entire genome of chloroplast in this study produced a well-resolved tree. But the relationship of the species in the V. dilatatum complex remains unclear. Given that the overall level of variations among the species in the V. dilatatum complex is low, the chloroplast genome may not provide phylogenetic signal for the reconstruction of phylogenetic relationship of the species. The four species in the V. dilatatum complex can be distinguished by morphological characters (Choi and Oh, 2019). Other molecular markers in the nuclear genome such as simple sequence repeats and SNP data may be needed to understand the phylogenetic relationship of the V. dilatatum complex.
Our analysis of chloroplast genome suggests that the genomic data are useful to infer phylogenetic relationships of Viburnum, producing the fully resolved tree with high bootstrap and posterior priority values (Fig. 4). The phylogenetic relationship of three clades, Succodontotinus, Lutescentia, and Lantana, was consistent with previous findings (Clement et al., 2014). Our analysis produced additional resolution within the Succodontotinus clade that was not resolved in previous phylogenetic analysis (Clement et al., 2014). Our analysis of entire chloroplast genome showed that members of the V. dilatatum complex formed a strongly supported clade and that V. betulifolium was sister to it. There are 400 SNPs between V. betulifolium and V. dilatatum, indicating a high degree of molecular divergence between the species at the genomic level. The results of phylogenetic analysis are consistent with the absence of gap region 1 in the V. dilatatum complex (Fig. 3B).