KIM, PARK, JANG, and SON: Characterization of the complete chloroplast genome sequence of Maesa japonica (Myrsinaceae)
Abstract
Maesa Forssk. (Myrsinaceae) has approximately 200 species distributed in the tropics from southern Africa to East Asia. Among them, Maesa japonica (Thunb.) Moritzi ex Zoll. is distributed throughout China, Japan, Taiwan, Vietnam, and Korea. In Korea, this species is found only on Jejudo Island. In this study, we conducted whole-genome sequencing of M. japonica chloroplast (cp). The complete cp genome sequence of M. japonica was found to be 157,189 bp in length, consisting of large (87,780 bp) and small (18,143 bp) single-copy regions (LSC and SSC, respectively) and a pair of identical inverted repeats (IRs) (25,633 bp). The overall GC content of the cp genome was 38%, while those of the LSC, SSC, and each IR were 35.2%, 30.9%, and 43.1%, respectively. Furthermore, 130 genes consisting of 85 protein-coding genes, 37 transfer RNA genes and eight ribosomal RNA genes were identified in the cp genome. A phylogenetic analysis of 12 taxa inferred from the cp genome revealed a close relationship between M. salicifolia and M. japonica. The complete cp genome sequence of M. japonica provides valuable information for future evolutionary and phylogenetic studies of Maesa.
Keywords: Maesa japonica, Myrsinaceae, phylogenetic relationship, plastome
INTRODUCTION
The genus Maesa Forssk. (Myrsinaceae) comprises approximately 200 species of woody shrubs, trees, scramblers, or lianas that are mainly distributed in the tropics, from southern Africa to east Asia ( Sumanon et al., 2021; Li et al., 2022; Larson et al., 2023). Among them, Maesa japonica (Thunb.) Moritzi ex Zoll. is distributed in Vietnam, China, Taiwan, Japan, and Korea; notably, in Korea, this species is found only on Jejudo Island ( Kim and Kim, 2011). Maesa japonica can be distinguished from related taxa by their leaf blade, which is lanceolate, oblong, rarely ovate, smooth, and flat, with subentire to distally serrate margins ( Chen and Pipoly, 1996). The chloroplast (cp) is an intracellular organelle that plays an essential role in photosynthesis and other primary and secondary metabolic processes ( Shinozaki et al., 1986; Brunkard et al., 2015). Chloroplasts have their genetic replication mechanisms and are inherited maternally in most angiosperms and gymnosperms; however, they are paternally inherited in some gymnosperms ( Yang et al., 2021). Typically, most cp genomes are circular 120–170 kb long DNA molecules with a quadripartite structure and consist of 120–130 genes, including a large single-copy (LSC, 80–90 kb), a small single-copy (SSC, 16–27 kb), and a pair of inverted repeats (IRs) ( Chumley et al., 2006; Cui et al., 2019; Yoo et al., 2021). Owing to its highly conserved sequence and structure, the cp genome is useful for phylogenetic and evolutionary studies of plants ( Palmer et al., 1988; Kwon et al., 2022). Recently, whole plastid genomes have been widely used in phylogenetic studies to analyze groups of plants whose evolutionary relationships remain unresolved ( Gitzendanner et al., 2018). In this study, we determined the complete cp genome sequences of an M. japonica individual from Korea and investigated the phylogenetic position of M. japonica within Myrsinaceae by analyzing our novel cp genome information with previously reported cp genomes of species in the family.
MATERIALS AND METHODS
A specimen of M. japonica was sampled from Cheongsuri, Hangyeong-myeon, Jeju-si, Jeju-do, Korea (33°18′08.7″N, 126°16′36.2″E; 158.5 m). The collected plant material was stored at the Korea National Arboretum Herbarium under the voucher number Cheongsuri-220427-001.
DNA was isolated from silica-dried leaves using a DNeasy Plant kit (Qiagen, USA). Subsequently, the extracted DNA was verified using 2% agarose gel electrophoresis, and a DNA library was prepared using a TruSeq Nano DNA Kit according to the protocol specified in the Sample Preparation Guide provided by the manufacturer (Macrogen Inc., Seoul, Korea). Genome paired-end sequencing was performed at Macrogen Inc. on an Illumina platform, MiSeq sequencer (Illumina Inc., San Diego, CA, USA), based on a 301-bp read size. A total of 7,439,526 paired-end reads were obtained, of which 115,105 reads (1.5%) were identified as cp genome sequences.
The complete cp genome was assembled using Geneious v9.0.5 (Biomatters, Auckland, New Zealand) and annotated using the GeSeq tool ( Tillich et al., 2017) and Geneious v9.0.5 (Biomatters). A circular map of the M. japonica cp genome was constructed using CPGview available at http://www.1 kmpg.cn/cpgview ( Liu et al., 2023). To infer the phylogenetic relationships of M. japonica with other species within Myrsinaceae, the complete cp genome sequences of 11 species were downloaded from the NCBI GenBank ( https://www.ncbi.nlm.nih.gov/genbank/). Primula farinosa var. koreana T. Yamaz. was used as an outgroup. Phylogenetic analysis was performed for 88 coding sequences of Maesa and related species. Alignments were performed using MAFFT v7.450 in Geneious v9.0.5 (Biomatters) ( Katoh et al., 2002; Katoh and Standley, 2013). A maximum likelihood (ML) bootstrap analysis with 1,000 replicates was performed with the best-fit model (TVM+F+I+G4) determined using the IQ-tree web server ( Trifinopoulos et al., 2016). Bayesian inference (BI) analysis was conducted using MrBayes v3.2.6 ( Ronquist et al., 2012) implemented in PhyloSuite ( Zhang et al., 2020). The best molecular evolution model for the BI analysis with the cp genome-dataset (GTR+F+I+G4) was obtained using ModelFinder v2.0 ( Kalyaanamoorthy et al., 2017).
RESULTS AND DISCUSSION
The complete cp genome sequence of M. japonica is 157,189 bp in length, with the cp reads showing an average coverage depth of 228.62. The sequence was deposited in GenBank under the accession number OQ759651. The LSC, SSC, and IRs of the cp genome sequence were 87,780 bp, 18,143 bp, and 35,633 bp, respectively. The overall GC content of the cp genome was 37.3%, while those of the LSC, SSC, and each IR were 35.2%, 30.9%, and 43.1%, respectively. The cp genome contained 130 unique genes, including 85 coding genes, eight ribosomal RNA (rRNA) genes, and 37 transfer RNA (tRNA) genes ( Fig. 1). Seventeen genes (six coding genes, four tRNA, and seven rRNA) were duplicated in the IRs ( Table 1). Nine protein-coding genes contained one intron, whereas two protein-coding genes contained two introns. Additionally, rps12 was identified as a trans-spliced gene. Sequences from 12 species including Primula farinosa var. koreana as an outgroup were used to infer ML- and BI-based phylogenies, both of which showed the same topology and high support for each branch ( Fig. 2). Phylogenetic analysis revealed that the Maesa is monophyletic and is a sister group to other taxa within Myrsinaceae. Additionally, M. japonica was found to be closely related to M. salicifolia E. Walker (MW801082.1). To the best of our knowledge, this is the first study to assemble and annotate the complete cp genome sequence of Maesa japonica. Originally, the generic limits and alignments in Myrsinaceae s.str. clade are rather unclear ( Yan et al., 2019). In our study, the phylogenetic tree is shown to contain two main subclades, and the topology of tree is almost identical with previous phylogenetic study based on 78 protein-coding genes ( Yan et al., 2019). Additionally, phylogenetic tree analysis revealed that the Maesa is a monophyletic group and that M. japonica is closely related to M. salicifolia, showing a high percentage (99.9%) of pairwise identity with identical gene order and contents. M. salicifolia is morphologically distinct from M. japonica by its leaf blade, which is linear to lanceolate or bullate, with revolute or entire margins; fruits are reddish, punctate-lineate, and wrinkled ( Chen and Pipoly, 1996). However, only a few cp genome sequences of Maesa were reported, further studies are needed to understand fully resolved evolutionary and phylogenetic relationships within the genus. The cp genome sequence of M. japonica provides useful information for future studies aimed at understanding the phylogenetic and evolutionary relationships among related species.
ACKNOWLEDGMENTS
This study was supported by a grant from ‘Silvics of Korea’ [KNA-1-1-18, 15-3], funded by the Korea National Arboretum.
Fig. 1.
Gene map of the complete cp genome of Maesa japonica. Genes within the circle are transcribed in a clockwise direction, whereas those drawn outside the circle are transcribed in a counterclockwise direction. Different gene colors correspond to different gene functions. The red and green arcs from the inner parts depict the dispersed repeats connected by forward direction and reverse direction, respectively. The short blue bars show the long tandem repeats. The purple areas indicate the extent of the inverted repeats, which separate the genome into large single-copy (LSC) and small single-copy (SSC) regions. 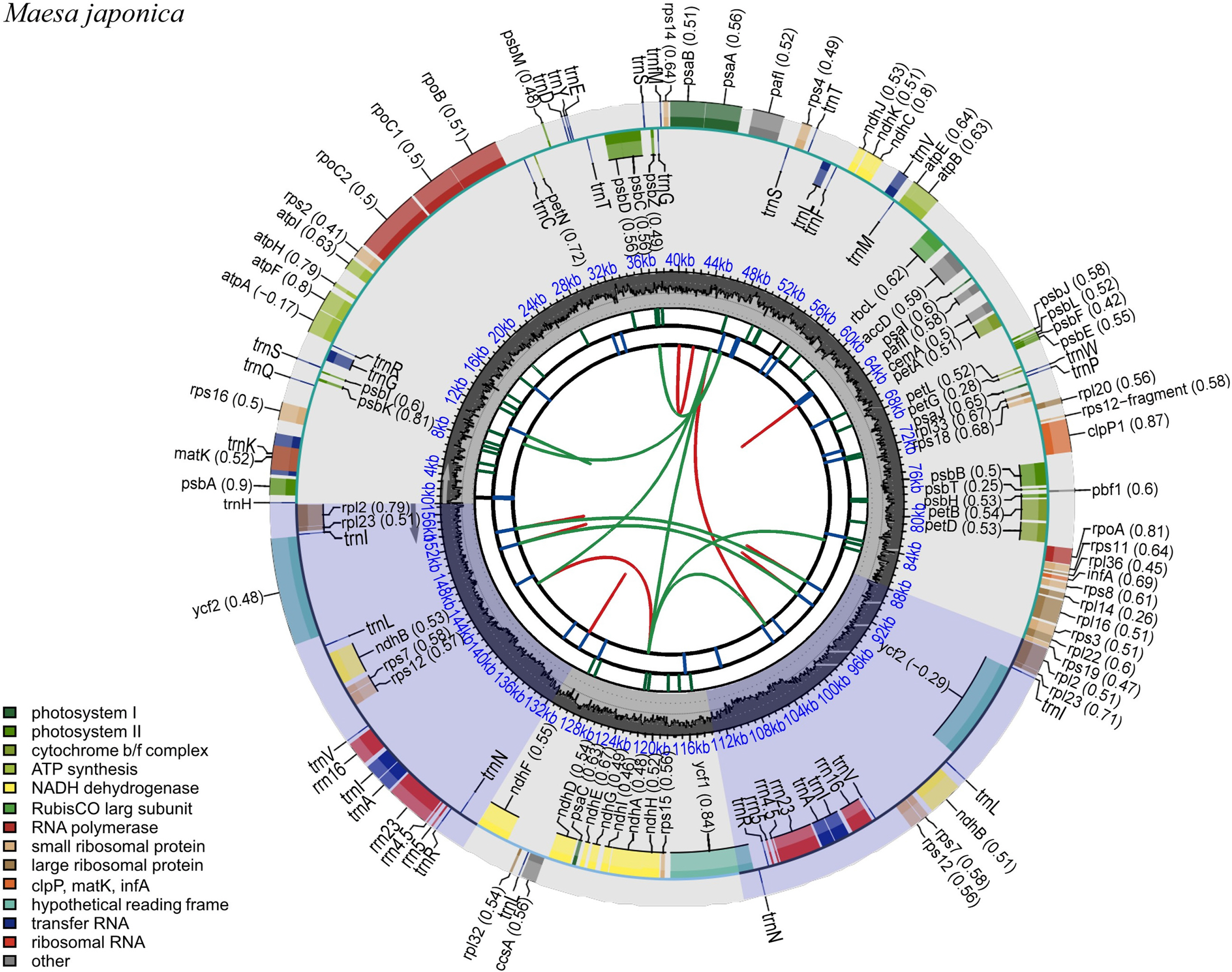
Fig. 2.
Phylogenetic tree of Maesa japonica and related taxa based on 88 protein-coding gene sequences inferred from Bayesian inference (BI) and maximum likelihood (ML) analysis. Numbers on each branch represent posterior probability values/bootstrap values. Primula farinosa var. koreana was used as an outgroup. 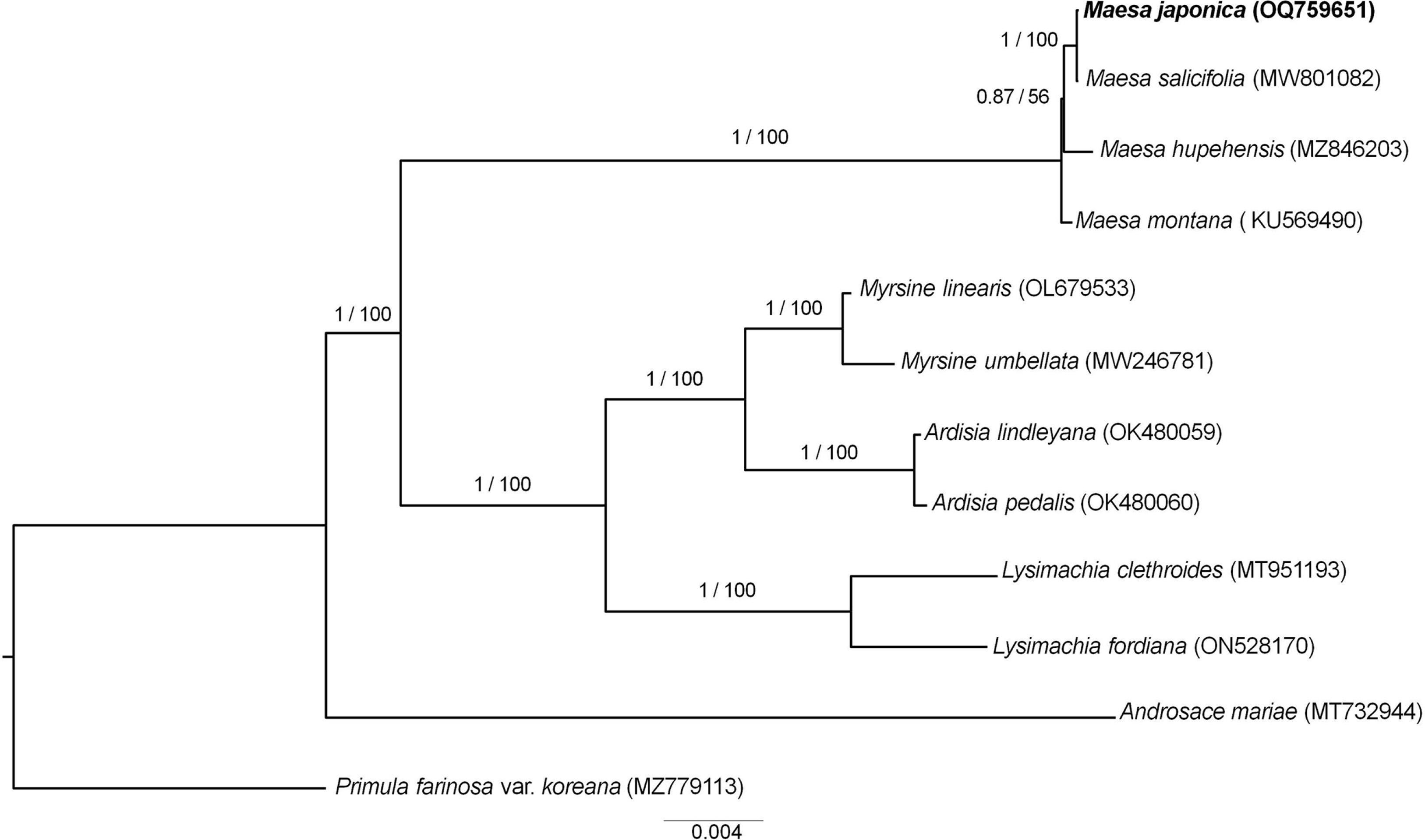
Table 1.
List of genes annotated in the chloroplast genome of Maesa japonica.
|
Category for genes |
Group of genes |
Name of genes |
|
Self-replication |
Large subunit ribosomal proteins |
rpl2(×2)*, rpl14, rpl16*, rpl20, rpl22, rpl23(×2), rpl32, rpl33, rpl36
|
|
DNA-dependent RNA polymerase |
rpoA, rpoB, rpoC1*, rpoC2
|
|
Small subunit ribosomal proteins |
rps2, rps3, rps4, rps7(×2), rps8, rps11, rps12(×2)T , rps14, rps15, rps16*, rps18, rps19
|
|
Ribosomal RNAs |
rrn4.5S(×2), rrn5S(×2), rrn16S(×2), rrn23S(×2) |
|
Transfer RNAs |
trnA-UGC(×2)*, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC*, trnH-GUG, trnI-CAU(×2), trnI-GAU(×2)*, trnK-UUU*, trnL-CAA(×2), trnL-UAA*, trnL-UAG, trnM-CAU, trnN-GUU(×2), trnP-UGG, trnQ-UUG, trnR-ACG(×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(×2), trnV-UAC*, trnW-CCA, trnY-GUA
|
|
Photosynthesis |
Subunits of ATP synthase |
atpA, atpB, atpE, atpF*, atpH, atpI
|
|
Subunits of NADH-dehydrogenase |
ndhA*, ndhB(×2)*, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK
|
|
Subunits of cytochrome b/f complex |
petA, petB*, petD*, petG, petL, petN
|
|
Subunits of photosystem I |
psaA, psaB, psaC, psaI, psaJ
|
|
Subunits of photosystem II |
psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbT, psbZ
|
|
Subunit of Rubisco |
rbcL
|
|
Photosystem assembly factors |
pafI**, pafII
|
|
Photosystem biogenesis factor |
pbf1
|
|
Other genes |
Subunit of Acetyl-CoA-carboxylase |
accD
|
|
C-type cytochrome synthesis gene |
ccsA
|
|
Envelop membrane protein |
cemA
|
|
ATP-dependent protease subunit P |
clpP**
|
|
Translational initiation factor |
infA
|
|
Maturase |
matK
|
|
Unknown function |
Conserved open reading frames |
ycf1, ycf2(×2) |
LITERATURE CITED
Chen, C and Pipoly, J. J. 1996. Myrsinaceae. Flora of China. Vol. 15. Myrsinaceae through Loganiaceae. Wu, Z. Y. Raven, P. H (eds.), Science Press, Beijing and Missouri Botanical Garden Press, St. Louis, MO. 1-38.
Chumley, T. W. Palmer, J. D. Mower, J. P. Fourcade, H. M. Calie, P. J. Boore, J. L and Jansen, R. K. 2006. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Molecular Biology and Evolution 23: 2175-2190.   Cui, Y. Chen, X. Nie, X. Sun, W. Hu, H. Lin, Y. Li, H. Zheng, X. Song, J and Yao, H. 2019. Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. International Journal of Molecular Sciences 20: 4040.  Gitzendanner, M. A. Soltis, P. S. Yi, T.-S. Li, D.-Z and Soltis, D. E. 2018. Plastome phylogenetics: 30 years of inferences into plant evolution. Advances in Botanical Research. Chaw, S.- M. Jansen, R. K (eds.), Academic Press, London. 293-313. Kim, J. S and Kim, T. Y. 2011. Woody Plants of Korean Peninsula. Dolbegea. Paju, 300 pp.
Kwon, S.-H. Park, Y. Jang, Y. L and Kwon, H.-Y. 2022. The complete chloroplast genome sequence of Hibiscus sabdariffa (Malvaceae). Korean Journal of Plant Taxonomy 52: 123-126.  Larson, D. A. Chanderbali, A. S. Maurin, O. Gonçalves, D. J. P. Dick, C. W. Soltis, D. E. Soltis, P. S. Fritsch, P. W. Clarkson, J. J. Grall, A. Davies, N. M. J. Larridon, I. Kikuchi, I. A. B. S. Forest, F. Baker, W. J. Smith, S. A and Utteridge, T. M. A. 2023. The phylogeny and global biogeography of Primulaceae based on high-throughput DNA sequence data. Molecular Phylogenetics and Evolution 182: 107702. Li, Y. Yao, X. Chang, Q. Zhang, C and Xia, P. 2022. Characterization of the complete chloroplast genome sequence of Maesa hupehensis Rehd (primulaceae). Mitochondrial DNA Part B Resources 7: 948-949. Palmer, J. D. Jansen, R. K. Michaels, H. J. Chase, M. W and Manhart, J. R. 1988. Chloroplast DNA variation and plant phylogeny. Annals of the Missouri Botanical Garden 75: 1180-1206. Ronquist, F. Teslenko, M. van der Mark, P. Ayres, D. L. Darling, A. Höhna, S. Larget, B. Liu, L. Suchard, M. A and Huelsenbeck, J. P. 2012. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539-542. Shinozaki, K. Ohme, M. Tanaka, M. Wakasugi, T. Hayashida, N. Matsubayashi, T. Zaita, N. Chunwongse, J. Okokata, J. Yamaguchi-Shinozaki, K. Ohto, C. Torazawa, K. Meng, B. Y. Sugita, M. Deno, H. Kamogashira, T. Yamada, K. Kusuda, J. Takaiwa, F. Kato, A. Tohdoh, N. Shimada, H and Sugiura, M. 1986. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. The EMBO Journal 5: 2043-2049. Sumanon, P. Eiserhardt, W. L. Balslev, H and Utteridge, T. M. A. 2021. Six new species of Maesa (Primulaceae) from Papua New Guinea. Phytotaxa 505: 245-261. Yang, X. Zhou, T. Su, X. Wang, G. Zhang, X. Guo, Q and Cao, F. 2021. Structural characterization and comparative analysis of the chloroplast genome of Ginkgo biloba and other gymnosperms. Journal of Forestry Research 32: 765-778. Yoo, S.-C. Oh, S.-H and Park, J. 2021. Phylogenetic position of Daphne genkwa inferred from complete chloroplast data. Korean Journal of Plant Taxonomy 51: 171-175.
Zhang, T. Zeng, C.-X. Yang, J.-B. Li, H.-T and Li, D.-Z. 2016. Fifteen novel universal primer pairs for sequencing whole chloroplast genomes and a primer pair for nuclear ribosomal DNAs. Journal of Systematics and Evolution 54: 219-227.
|
|











