ZOU, BAI, XIANG, and LI: Characterization of the complete chloroplast genome sequence of Hypericum petiolulatum (Hypericaceae)
Abstract
Hypericum L. (Hypericaceae) is one of the best-selling herbal medicines in the world comprising ca. 500 species of herbs, shrubs, and small trees. Hypericum petiolulatum Hook. f. & Thomson ex Dyer is widely distributed in China, Vietnam, Myanmar, Nepal, India, Malaysia, and Bhutan and is used as a traditional herb to treat hemoptysis and inflammation. In this study, we sequenced and assembled the complete chloroplast (cp) genome of H. petiolulatum. The complete plastome of H. petiolulatum was 136,105 bp in length, with a large single copy region (LSC) of 93,709 bp, a small single copy region (SSC) of 11088 bp and two identical inverted repeats (IRs) of 15,654 bp. The overall GC content of the plastome was 37.0%, while GC contents of the LSC, SSC, and each IR were 35.5%, 31.0%, and 43.8%, respectively. In addition, 116 genes consisting of 76 protein-coding genes, six ribosomal RNA genes, and 34 transfer RNA genes were identified. A phylogenetic analysis of 14 taxa inferred based on cp genome sequences revealed a close relationship between H. petiolulatum and H. perforatum. The complete cp genome sequence of H. petiolulatum reported in this paper will facilitate population and phylogenomics studies of this medicinal plant group.
Keywords: chloroplast genome, Hypericaceae, Hypericum petiolulatum, phylogenetic relationship
INTRODUCTION
Hypericum L. is the largest genus within Hypericaceae, comprising at least 500 species of herbs, shrubs, and infrequently small trees ( Robson, 1981, 2012, 2016; The Angiosperm Phylogeny Group, 2016). The genus has a near-cosmopolitan distribution, with Eurasia (ca. 230 spp.) and Andean South America (ca. 130 spp.) are the major diversity centers ( Meseguer et al., 2013; Nürk et al., 2013). A lot of species has been used as traditional medicine in the world, such as H. perforatum L. (st. John’s wort) is widely used in different countries to treat mild to moderate mental depression. Hypericum petiolulatum Hook.f. & Thomson ex Dyer is another medicinal plant widely distributed in China, Vietnam, Myanmar, Nepal, India, Malaysia, and Bhutan ( Robson, 1981; Li and Robson, 2007). This species is an ethnomedicines used to treat hemoptysis and inflammation ( Zhang et al., 2020b). Morphologically, this species can be distinguished from related taxa by the longer petiole, shorter styles, broader capsule, and leaves broadest at the above middle ( Li and Robson, 2007).
Although chloroplast (cp) genomes have a low substitution rate than nuclear genomes, compared with nuclear genomes, cp genomes are highly conserved in gene composition, variation ratio, and structure ( Dobrogojski et al., 2020). Currently, cp genomes are often used for phylogenetic studies at different taxonomic levels ( Xiang et al., 2019; Zhao et al., 2021; Dong et al., 2022; Zhao et al., 2023).
Hypericum is a species-diverse genus with considerable medicinal and ornamental value, however, plastome of this genus are rarely reported in comparison with other large genus, such as Rhododendron L., Salvia L., and Solanum L. ( Daniell et al., 2006; Zhao et al., 2020; Shirasawaet al., 2021). To date, plastome of only five species have been published and therefore little is known regarding plastome structure variation within Hypercium.
In this study, we sequenced and assembled the plastome of H. petiolulatum, which is widely distributed and medicinally used in Asia. In addition, 12 previously published plastomes of Hypericaceae were included for phylogenetic analyses to infer the phylogenetic relationships of H. petiolulatum and related taxa.
MATERIALS AND METHODS
Fresh leaves of Hypericum petiolulatum were collected from Guangyuan, Sichuan, southwest China (32°37′56.34″N, 106°15′20.57″E; 1,677 m). Voucher specimen (XCL 2488) was deposited in Kunming Institute of Botany, Chinese Academy of Sciences.
Total genomic DNA (gDNA) was isolated using the modified CTAB method ( Doyle and Doyle, 1987) from silicagel-dried leaf material. Subsequently, the gDNA were quantified by Tiangen DNA secure Plant Kit (DP320) and DNA concentration was detected using NanoDrop spectrophotometer 2000 (Thermo Scientific, Carlsbad, CA, USA) to ensure that the DNA concentration used for library construction was greater than 30 ng/μL. The gDNA was sheared into fragments of about 300 bp to construct libraries using the NEBNext Ultra II DNA Library Prep Kit for Illumina. Genome paired-end sequencing was performed on the Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA) at BGI Genomics (BGI-Shenzhen, Shenzhen, China). Approximately 5 GB of raw data was generated with 150 bp paired-end read lengths. Quality control of raw sequence reads was carried out using “fastp” with the default parameter ( Chen et al., 2018). The complete cp genome was assembled using the “GetOrganelle” pipeline ( Jin et al., 2020) and annotated using the “Plastid Genome Annotator (PGA)” ( Qu et al., 2019) with the published plastome of Amborella trichopoda Baill. (AJ506156) ( Goremykin et al., 2003) as a reference. The putative start and stop codons of protein-coding genes, and intron/exon positions were manually adjusted in “Geneious version 9.0.2” ( Kearse et al., 2012). A circular map of the plastome of H. petiolulatum was obtained using “CPGview” ( Liu et al., 2023). To infer the phylogenetic relationships of H. petiolulatum and related taxa within Hypericum, the complete cp genome sequences of 14 species were downloaded from the NCBI GenBank ( https://www.ncbi.nlm.nih.gov/genbank/), including two species of Cladopus H.A. Möller were selected as outgroup. The script “get_annotated_regions_from_gb.py” develop by Zhang et al. (2020c) was used to extracted 68 protein-coding genes (CDS) for phylogenetic analysis. Alignment was firstly performed with “MAFFT v7.310” ( Katoh and Standley, 2013) and then “TrimAl v.1.4.1” ( Capella-Gutiérrez et al., 2009) was used for automated alignment trimming. A concatenated sequence matrix was generated using “Phylosuite v.1.2.2” ( Zhang et al., 2020a). Phylogenetic analysis was conducted using maximum likelihood (ML), which was method implemented on the “Cyberinfrastructure for Phylogenetic Research Science (CIPRES) Gateway v.3.3 server” ( Miller et al., 2010), the GTRCAT model was selected, bootstrap probabilities were generated by conducting 1,000 reiterations, and other parameters used the CIPRES default settings. The resulting tree with nodal support values was visualized and edited using “FigTree v.1.4.3”( http://tree.bio.ed.ac.uk/software/figtree/).
RESULTS AND DISCUSSION
The annotated plastid genome sequence has been submitted to the GenBank with the accession number PP085178. The plastome displayed the typical quadripartite structure ( Fig. 1). The whole plastid genome of H. petiolulatum was 136,105 bp in length, which consisted of two inverted repeats (IRs) of 15,654 bp that separated a large single copy (LSC) of 93,709 bp and a small single copy (SSC) of 11,088 bp. The overall GC content of the plastome was 37.0%, the LSC, SSC, and each IR were 35.5%, 31.0%, and 43.8%, respectively. Plastome of H. petiolulatum consist of a total of 116 genes, including 76 coding genes (CDS), six ribosomal rRNA genes, and 34 tRNA genes ( Table 1). The ML analysis showed that Hypericaceae was well supported as monophyly and consisted of two large clades, i.e., Hypericum and Cratoxylum Blume ( Fig. 2). Species of two genera formed two monophyletic clades, respectively. Within Hypericum, H. breviflorum (Wall. Ex Dyer) Y. Kimura, followed by H. perforatum + H. petiolulatum and then H. ascyron L. + H. monogymum L. and H. hookerianum Wight & Arn., which in turn formed sister group and fully supported (100%). Although only six species representing five sections were included for phylogenetic analysis, relationship among those sections is comparable with previous studies ( Crockett et al., 2004; Pilepić et al., 2010). For example, the close relationship between H. ascyron and H. hookerianum was reported by Pilepić et al. (2010) and here we recovered the relationship. In addition, there are morphological correlates with the phylogenetic relationships. Here, we found that H. hookerianum, H. monogymum, and H. ascyron formed a subclade, some morphological characters also support this relationship, i.e., seeds of those species have a conspicuous winglet at the apex and are carinate in lateral margins ( Bai et al., 2023), ovary with five styles, and black glands absent in leaves and sepals ( Li and Robson, 2007). The focal species, H. petiolulatum was found to be closely related to H. perforatum L., both species have dense or irregular black glands on the leaves, sepals, and anthers ( Li and Robson, 2007).
Hypericum is a taxonomically difficult group and species relationships were not well resolved in all previous studies ( Pilepić et al., 2010; Meseguer et al., 2013; Nürk et al., 2013). In total, only five DNA markers ( psbA- trnH, trnL-trnF, trnS- trnG, rbcL, and internal transcribed spacer) were used in these studies and not generated phylogenetic trees with high resolution, the part reason is due to lacking of variability within these DNA markers. Despite the limited sampling, this study provides a better supported phylogeny than previous studies ( Crockett et al., 2004; Pilepić et al., 2010), indicating complete plastome sequences can markedly improve phylogenetic resolution. Therefore, we can expect that complete plastome sequences based on large-scale sampling can further resolve species relationships within the genus and contribute to a better understanding of the infrageneric relationships of Hypericum.
ACKNOWLEDGMENTS
We gratefully acknowledge the support provided by the National Natural Science Foundation of China to CLX (No. 32370221), the CAS Interdisciplinary Team of the “Light of West China” Program and Yunnan Revitalization Talent Support Program “Innovation Team” Project.
Fig. 1.
The circular map and gene structure of the plastome of Hypericum petiolulatum. Genes inside and outside the circle are transcribed in the clockwise and counterclockwise directions, respectively. Different gene colors correspond to different gene functions. The red and green arcs in the inner parts depict the dispersed repeats connected by forward and reverse direction, respectively. The short blue bars show the tandem repeats. The purple areas indicate the extent of the inverted repeats (IRa and IRb), which separate the genome into large single-copy (LSC) and small single-copy (SSC) regions. The overall GC content of the plastome was 37.0%, the LSC, SSC, and each IR were 35.5%, 31.0%, and 43.8%, respectively. 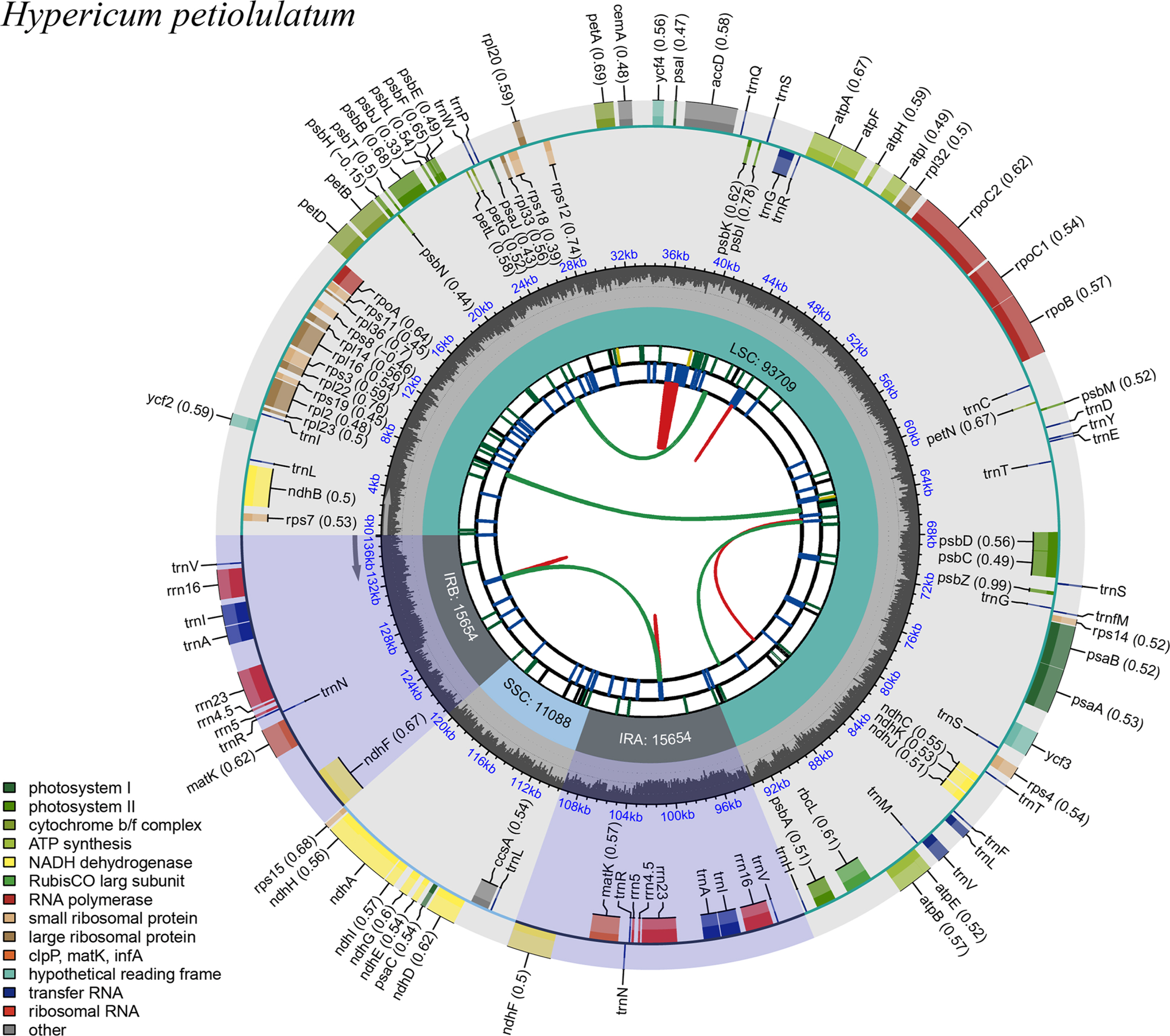
Fig. 2.
Maximum likelihood analysis based a combined dataset of 68 protein-coding genes. Numbers on each branch represent bootstrap support values (BS). 
Table 1.
List of genes annotated in the chloroplast genome of Hypericum petiolulatum.
|
Category for genes |
Function of genes |
Names of genes |
|
Self-replication |
Large subunit ribosomal proteins |
rpl36, rpl33, rpl32, rpl23, rpl22, rpl20, rpl16*, rpl14, rpl2*
|
|
DNA-dependent RNA polymerase |
rpoC2, rpoC1, rpoB, rpoA
|
|
Small subunit ribosomal proteins |
rps19, rps18, rps15, rps14, rps12*T, rps11, rps8, rps7, rps4, rps3
|
|
Ribosomal RNAs |
rrn23, rrn16(×2), rrn5(×2), rrn4.5
|
|
Transfer RNAs |
trnY-GUA, trnW-CCA, trnV-UAC*, trnV-GAC(x2), trnT-UGU, trnT-GGU, trnS-UGA, trnS-GGA, trnS-GCU, trnR-UCU, trnR-ACG(×2), trnQ-UUG, trnP-UGG, trnN-GUU(×2), trnM-CAU, trnL-UAG, trnL-UAA*, trnL-CAA, trnI-GAU(×2)*, trnI-CAU, trnH-GUG, trnG-UCC*, trnG-GCC, trnfM-CAU, trnF-GAA, trnE-UUC, trnD-GUC, trnC-GCA, trnA-UGC(×2)*
|
|
Photosynthesis |
Subunits of ATP synthase |
atpI, atpF*, atpH, atpE, atpB, atpA
|
|
Subunits of NADH-dehydrogenase |
ndhK, ndhJ, ndhI, ndhH, ndhG, ndhF(×2), ndhE, ndhD, ndhC, ndhB*, ndhA*
|
|
Subunits of cytochrome b/f complex |
petN, petL, petG, petD*, petB*, petA
|
|
Subunits of photosystem I |
psaJ, psaI, psaC, psaB, psaA
|
|
Subunits of photosystem II |
psbZ, psbT, psbN, psbM, psbL, psbK, psbJ, psbI, psbH, psbF, psbE, psbD, psbC, psbB, psbA
|
|
Subunit of Rubisco |
rbcL |
|
Other genes |
Subunit of acetyl-CoA-carboxylase |
accD |
|
C-type cytochrome synthesis gene |
ccsA |
|
Envelop membrane protein |
cemA |
|
Maturase |
matK(×2) |
|
Unknown function |
Conserved open reading frames |
ycf4, ycf3*, ycf2 |
LITERATURE CITED
Bai, R.-Z., Zhao, F. Drew, B. T. Xu, G. X. Cai, J. Shen, S.-K. and Xiang, C.-L. 2023. Seed morphology of Hypericum (Hypericaceae) in China and its taxonomic significance. Microscopy Research and Technique 86: 1496-1509.   Crockett, S. L., Douglas, A. W. Scheffler, B. E. and Khan, I. A. 2004. Genetic profiling of Hypericum (St. John’s Wort) species by nuclear ribosomal ITS sequence analysis. Planta Medica 70: 929-935. Daniell, H., Lee, S.-B. Grevich, J. Saski, C. Quesada-Vargas, T. Guda, C. Tomkins, J. and Jansen, R. K. 2006. Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes. Theoretical and Applied Genetics 112: 1503-1518.  Dobrogojski, J., Adamiec, M. and Luciński, R. 2020. The chloroplast genome: a review. Acta Physiologiae Plantarum 42: 98. Doyle, J. J. and Doyle, J. L. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11-15.
Goremykin, V. V., Hirsch-Ernst, K. I. Wölfl, S. and Hellwig, F. H. 2003. Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal angiosperm. Molecular Biology and Evolution 20: 1499-1505. Katoh, K. and Standley, D. M. 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biological and Evolution 30: 772-780. Kearse, M., Moir, R. Wilson, A. Stones-Havas, S. Cheung, M. Sturrock, S. Buxton, S. Cooper, A. Markowitz, S. Duran, C. Thierer, T. Ashton, B. Meintjes, P. and Drummond, A. 2012. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647-1649.  Li, X.-W. and Robson, N. K. B. 2007. Hypericum. In Flora of China. Vol. 13. Clusiaceae. Wu, Z. Y., Raven, P. H. and Hong, D. Y. (eds.), Science Press, Beijing and Missouri Botanical Garden Press, St. Louis, MO. Pp. 2-35.
Meseguer, A. S., Aldasoro, J. J. and Sanmartín, I. 2013. Bayesian inference of phylogeny, morphology and range evolution reveals a complex evolutionary history in St. John's wort ( Hypericum). Molecular Phylogenetics and Evolution 67: 379-403. Miller, M. A., Pfeiffer, W. and Schwartz, T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, Louisiana. 14 Nov 2010. Piscataway: IEEE: 45-52. Nürk, N. M., Madriñán, S. Carine, M. A. Chase, M. W. and Blattner, F. R. 2013. Molecular phylogenetics and morphological evolution of St. John's wort ( Hypericum; Hypericaceae). Molecular Phylogenetics and Evolution 66: 1-16. Pilepić, K. H., Morović, M. Orač, F. Šantor, M. and Vejnović, V. 2010. RFLP analysis of cpDNA in the genus Hypericum. Biologia 65: 805-812. Robson, N. K. B. 1981. Studies in the genus Hypericum L. (Guttiferae). 2. Characters of the genus. Bulletin of the British Museum (Natural History) Botany 8: 55-226.
Robson, N. K. B. 2012. Studies in the genus Hypericum L. (Hypericaceae) 9. Addenda, corrigenda, keys, lists and general discussion. Phytotaxa 72: 1-111. Robson, N. K. B. 2016. And then came molecular phylogenetics: Reactions to a monographic study of Hypericum (Hypericaceae). Phytotaxa 255: 181-198. Shirasawa, K., Kobayashi, N. Nakatsuka, A. Ohta, H. and Isobe, S. 2021. Whole-genome sequencing and analysis of two azaleas, Rhododendron ripense and Rhododendron kiyosumense. DNA Research 28: dsab010. The Angiosperm Phylogeny Group, Chase, M. W. Christenhusz, M. J. M. Fay, M. F. Byng, J. W. Judd, W. S. Soltis, D. E. Mabberley, D. J. Sennikov, A. N. Soltis, P. S. and Stevens, P. F. 2016. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society 181: 1-20. Xiang, C.-L., Dong, H.-J. Landrein, S. Zhao, F. Yu, W.-B. Soltis, D. E. Soltis, P. S. Backlund, A. Wang, H.-F. Li, D.-Z. and Peng, H. 2019. Revisiting the phylogeny of Dipsacales: New insights from phylogenomic analyses of complete plastomic sequences. Journal of Systematics and Evolution 58: 103-117. Zhang, R., Ji, Y. Zhang, X. Kennelly, E. J. and Long, C. 2020b. Ethnopharmacology of Hypericum species in China: A comprehensive review on ethnobotany, phytochemistry and pharmacology. Journal of Ethnopharmacology 254: 112686. Zhang, R., Wang, Y.-H. Jin, J.-J. Stull, G. W. Bruneau, A. Cardoso, D. De Queiroz, L. P. Moore, M. J. Zhang, S.-D. Chen, S.-Y. Wang, J. Li, D.-Z. and Yi, T.-S. 2020c. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Systematic Biology 69: 613-622. Zhao, F., Chen, Y.-P. Salmaki, Y. Drew, B. T. Wilson, T. C. Scheen, A.-C. Celep, F. Bräuchler, C. Bendiksby, M. Wang, Q. Min, D.-Z. Peng, H. Olmstead, R. G. Li, B. and Xiang, C.-L. 2021. An updated tribal classification of Lamiaceae based on plastome phylogenomics. BMC Biology 19: 2. Zhao, F., Li, B. Drew, B. T. Chen, Y.-P. Wang, Q. Yu, W.-B. Liu, E.-D. Salmaki, Y. Peng, H. and Xiang, C.-L. 2020. Leveraging plastomes for comparative analysis and phylogenomic inference within Scutellarioideae (Lamiaceae). PLoS ONE 15: e0232602. Zhao, Y., Chen, Y.-P. Yuan, J.-C. Paton, A. J. Nuraliev, M. S. Zhao, F. Drew, B. T. Salmaki, Y. Turginov, O. T. Sun, M. Sennikov, A. N. Yu, X.-Q. Li, B. and Xiang, C.-L. 2023. Museomics in Lamiaceae: Resolving the taxonomic mystery of Pseudomarrubium. Current Plant Biology 35–36: 100300.
|
|











